Устойчивостьдисперсной системы характеризуется неизменностью во времени ее основных параметров: дисперсности и равновесного распределения дисперсной фазы в среде.
Проблема устойчивости - одна из самых важных и сложных в коллоидной химии, это проблема «жизни и смерти» дисперсной системы. Прежде всего, отметим резкое различие в отношении устойчивости между двумя основными классами: лиофильными и лиофобными коллоидами.
Лиофильные системы — молекулярные коллоиды, а также лиофильные суспензоиды (например, глины, мыла) диспергируются самопроизвольно, образуя термодинамически устойчивые коллоидные растворы; свободная энергия системы в этом процессе уменьшается:
 ,
,
где F - энергия Гельмгольца, U - внутренняя энергия, Т - абсолютная температура, S - энтропия.
Использование этого условия в качестве критерия лиофильности (П.А.Ребиндер и др.) позволяет провести вполне четкую количественную границу между двумя классами дисперсных систем.
Увеличение энтропии в процессе диспергирования обычно способствует уменьшению F, поскольку система приходит к более вероятному равномерному распределению дисперсной фазы в среде.
Баланс внутренней энергии ΔU в этом процессе складывается из затраты энергии на разрыв молекулярных связей с образованием новой поверхности (работы когезии) и выигрыша — в результате межфазного сольватационного взаимодействия (работы адгезии). Для лиофильных систем обычно ΔU < 0, энергия межфазного взаимодействия(Wа) значительно превышает энергию связи внутри дисперсной фазы.
Усиление взаимодействия дисперсной фазы со средой (лио-фильности) способствует самопроизвольному диспергированию.
Самопроизвольно диспергирующиеся системы (ΔF < 0) определяются как лиофильные.
Лиофобные коллоиды, наоборот, характеризуются энергией связи внутри дисперсной фазы (Wс), значительно большей, чем энергия межфазного взаимодействия (Wа), и эта разность не компенсируется энтропийным фактором; для них:
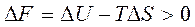 .
.
Диспергирование не может идти самопроизвольно и совершается либо за счет внешней работы, либо за счет других процессов, протекающих в системе спонтанно (например, химических). Образующиеся дисперсии термодинамически неустойчивы и характеризуются высокими значениями σ на межфазной границе, которая соответствует малой величине Wа.
Таким образом, системы, не диспергирующиеся самопроизвольно (ΔF > 0), определяются как лиофобные коллоиды, хотя и для них всегда характерна та или иная степень межфазного взаимодействия (лиофилизации).
Рассмотрим устойчивость лиофобныхколлоидов.
Несмотря на термодинамическую неустойчивость (ΔF > 0), многие лиофобные коллоидные системы оказываются вполне устойчивыми кинетически, не изменяясь заметно в течение длительного времени (иногда десятилетиями). Очевидно, что эти системы существуют в метастабильном состоянии, т.е. потенциальный барьер для них достаточно высок.
Для уяснения причин относительной устойчивости подобных систем следует определить прежде всего, о каком виде устойчивости идет речь. Понятие о различных видах устойчивости — седиментационной (кинетической) и агрегативной — было введено Н.П.Песковым.
Седиментационной называется устойчивость дисперсной фазы по отношению к силе тяжести. Нарушение ее и, как следствие, разрушение системы (разделение фаз), может быть вызвано:
- седиментацией частиц, характерной для грубодисперсных систем, приводящей к оседанию (или всплыванию) дисперсной фазы. Высокодисперсные системы — кинетически устойчивы; для них характерно установление седиментационного равновесия;
- изотермической перегонкой мелких частиц вещества дисперсной фазы в более крупные с последующей седиментацией;
- потерей агрегативной устойчивости в результате объединения частиц, приводящего к коагуляции дисперсной фазы.
Коагуляция — процесс слипания частиц, образования более крупных агрегатов с потерей седиментационной устойчивости и последующим разделением фаз — разрушением дисперсной системы.
Таким образом, агрегативная устойчивость может быть определена как способность системы к сохранению дисперсности и индивидуальности частиц дисперсной фазы. Образующиеся в результате потери агрегативной устойчивости коагуляты представляют собой осадки (или всплывающие образования) различной структуры — плотные, творожистые, хлопьевидные, волокнистые, кристаллоподобные.
В лиофобных системах структура коагулятов и их прочность в значительной мере определяется степенью сольватации, которая может изменяться в весьма широком диапазоне — от типично лиофобных коллоидов (гидрозоли металлов) до систем сильно лиофилизированных, особенно в результате адсорбции ПАВ или ВМС. В подобных агрегатах, несмотря на изменение подвижности, частицы еще сохраняются как таковые большее или меньшее время (называемое «временем жизни»), после чего могут срастаться (в случае твердой дисперсной фазы) или сливаться (в случае жидкой) самопроизвольно с уменьшением поверхности раздела фаз. Срастание происходит в результате кристаллизации или конденсации; слияние капелек называется коалесценцией. Наблюдаемая долговечность многих лиофобных систем свидетельствует о том, что наряду с ван-дер-ваальсовыми силами притяжения между частицами в системе существуют и силы отталкивания или эффекты, экранирующие притяжение.
Многочисленные экспериментальные данные показывают, что в таких кинетически устойчивых типично гидрофобных системах всегда наблюдается заметный электрофорез**. Прекращение его в силу тех или иных причин вызывает немедленную коагуляцию. Эта связь приводит к представлению об электрической природе сил отталкивания в типично гидрофобных системах, тем более, что увеличение концентрации электролита, уменьшающее, Ψd и ξ -потенциалы) всегда приводит к коагуляции.
В лиофилизированных и лиофильных системах явной связи между величиной ξ- потенциала и устойчивостью не наблюдается; коагуляция вызывается десольватирующими агентами и дисперсные системы оказываются тем более устойчивыми, чем сильнее развиты сольватные оболочки.
6.1 КОАГУЛЯЦИЯ ГИДРОФОБНЫХ КОЛЛОИДОВ
Рассмотрим важнейшие экспериментальные факты, способные служить базой для построения теории.
Многообразие причин, вызывающих коагуляцию. Едва ли существуют какие-либо внешние воздействия, которые при достаточной интенсивности и длительности не вызвали бы коагуляции. Действие тепла и холода, электромагнитных полей, жестких излучений, механические воздействия, химические агенты приводят к нарушению агрегативной устойчивости и, следовательно, к коагуляции. Все эти воздействия, столь различные по характеру, обладают общим свойством - они разрушают энергетический барьер, и метастабильная система переходит в более устойчивое состояние самопроизвольно в процессе коагуляции.
Коагулирующее действие электролитов. Все без исключения электролиты вызывают коагуляцию при увеличении их концентрации в растворе до некоторого (различного для разных электролитов) критического значения ск, называемого порогом коагуляции (или коагулирующей концентрацией, величиной коагуляции, концентрацией коагуляции).
Ранее было установлено, что порогу коагуляции соответствует некоторое критическое значение ξ ≈ 30 мВ, ниже которого наступает коагуляция. Более поздние работы, проведенные с учетом поправок, необходимых для правильного вычисления ξ, заставили усомниться в существовании столь простой связи между зарядом и устойчивостью. Поэтому в современных теориях ξ -потенциалу отводится более скромная роль, хотя параметры ДЭС продолжают сохранять основное значение для оценки устойчивости, в частности величина Ψd,- определяющая однозначно (при постоянной ионной силе) силу электростатического отталкивания. Расчет сил отталкивания не дает, однако, однозначного решения задачи определения границ устойчивости, поскольку эти силы всегда действуют на фоне сил притяжения.
Влияние величины заряда иона. Величины ск сильно различаются для различных электролитов. Многочисленные исследования, проведенные с ионными рядами (различными катионами с одним общим анионом и наоборот), показали, что величина ск определяется зарядом противоиона. Эти результаты подтвердили представление о решающей роли внешней обкладки в процессе коагуляции. Коагулирующая способность противоиона Vк, определяемая как Vк = 1/ск см3/моль-ион и выражаемая в объемах золя, скоагулированных 1 моль-ион, резко возрастает с увеличением заряда противоиона. Так, при переходе от однозарядного противоиона к трехзарядному коагулирующая способность увеличивается не в 3, а в большое количество раз.
Установленные закономерности нашли свое выражение в правиле Шульце - Гарди, высказанном еще в конце XIX в. Оно может быть сформулировано следующим образом: коагулирующее действие оказывает противоион, и коагулирующая способность возрастает пропорционально некоторой высокой степени его заряда.
6.2 КИНЕТИКА БЫСТРОЙ КОАГУЛЯЦИИ.
ТЕОРИЯ СМОЛУХОВСКОГО
Результаты исследования зависимости скорости коагуляции от концентрации электролита с показывают, что если с мало, то υ = 0. Далее, в узком интервале концентраций наблюдается быстрый рост υ до некоторой величины, не изменяющейся с дальнейшим увеличением с. В соответствии с этим можно выделить три четко разграниченных зоны: устойчивости, медленной коагуляции (с порогом ск.м) и быстрой коагуляции (с порогом ск.б).
Область быстрой коагуляции определяется как область, в которой все соударения эффективны. Вычисление υ для этой области существенно упрощается, поскольку сводится к подсчету числа столкновений. Однако и здесь возникает немало трудностей, так как приходится учитывать столкновения не только первичных частиц, но и более сложных (двойников, тройников и т. д.), образующихся в процессе коагуляции. Эта задача была решена М. Смолуховским (1916 г.), предложившим количественную трактовку кинетики быстрой коагуляции на основе рассмотрения броуновского движения (диффузии) частиц.
Скорость процесса - функция концентрации частиц ν и интенсивности броуновского движения, характеризуемой коэффициентом диффузии D.
 . (48)
. (48)
Период коагуляции τ, являющийся основным параметром процесса быстрой коагуляции, представляется, согласно уравнению (48), функцией D и R. Учитывая, что эти две величины взаимосвязаны посредством уравнения Эйнштейна: D = kT/6πηa и а = R/2, находим:
 .
.
Из этого выражения видно, что τ не зависит в действительности от размера частиц, являясь функцией лишь вязкости среды η, абсолютной температуры Т иначальной концентрации частиц ν0.
Медленная коагуляция может быть объяснена неполной эффективностью столкновений, вследствие существования энергетического барьера. При введении доли эффективных столкновений α (по Смолуховскому) все уравнения сохраняют прежний вид, за исключением выражения для τ (48), которое увеличивается в 1/α раз:
 .
.
Таким образом, кривые ν - t для быстрой и медленной коагуляции должны совпадать при изменении масштаба времени. Однако экспериментальные кривые в случае медленной коагуляции не показали линейной зависимости.
Существует более совершенная теория Н.А. Фукса, которая будет рассмотрена после ознакомления с количественной интерпретацией величин энергетических барьеров в теории устойчивости гидрофобных коллоидов.
6.3 ТЕОРИЯ УСТОЙЧИВОСТИ ГИДРОФОБНЫХ КОЛЛОИДОВ ДЛФО
В процессе развития коллоидной химии возникло немало теорий, пытавшихся связать устойчивость гидрофобных золей, в частности коагулирующее действие электролитов, с теми или иными параметрами системы и явлениями, возникающими при взаимодействии дисперсной фазы с дисперсионной средой.
Так, химическая теория (П.Дюкло) выдвигала в качестве причины коагуляции химические реакции на границе раздела фаз, приводящие к «нейтрализации» поверхностного заряда.
Адсорбционная теория (Г.Фрейндлих) объясняла снижение заряда процессом адсорбции ионов. Согласно электростатической теории (Ф.Мюллер), увеличение с приводит, при постоянном заряде, к снижению ξ-потенциала, следовательно, и устойчивости системы. Теория А.И.Рабиновича рассматривала совместное действие ионного обмена и снижение ξ - потенциала. В теории, развитой В.Оствальдом, коагуляция рассматривалась как «вытеснение» дисперсной фазы межионными силами притяжения, действующими в дисперсионной среде (сжатие динамической ионной «решетки»). В этом представлении параметром, определяющим коагуляцию, является величина коэффициента активности электролита.
Каждая из этих теорий объясняла ряд фактов, но оказывалась бессильной перед множеством других, поскольку эти теории были в значительной степени односторонними, связывая сложный процесс коагуляции с каким-либо одним параметром системы.
Этого недостатка лишена современная теория устойчивости, развитая Б.В.Дерягиным (1937—1941 гг.) совместно с Л.Д.Ландау и получившая всеобщее признание. Несколько позже теоретическая разработка, почти аналогичная и приводящая к тем же результатам, была осуществлена независимым путем Э.Фервеем и Я.Овербеком. Поэтому для современной теория устойчивости принято обозначение ДЛФО.
В своем классическом варианте теория рассматривает процесс коагуляции, как результат совместного действия ван-дер-ваальсовых сил притяжения и электростатических сил отталкивания между частицами. В зависимости от баланса этих сил в тонкой прослойке жидкости между сближающимися телами возникает либо положительное «расклинивающее» давление, препятствующее их соединению, либо отрицательное, - приводящее к утончению прослойки и образованию контакта между частицами.
Существование расклинивающего давления π, величина которого зависит от толщины прослойки, было установлено экспериментально методами сближения воздушного пузырька со стеклянной пластинкой или двух скрещенных нитей в растворе электролита. Оказалось, что π резко возрастает с уменьшением толщины прослойки Н и концентрации электролита.
6.5.1 МИЦЕЛЛООБРАЗОВАНИЕ И СОЛЮБИЛИЗАЦИЯ
Молекулы ПАВ в водных растворах способны к образованию мицелл при достижении концентрации, называемой критической концентрацией мицеллобразования (ККМ). ККМ лежит обычно в пределах 10-3 - 10-6 моль/дм3. После достижения этой концентрации в растворе самопроизвольно образуются сферические мицеллы (мицеллы Гартли) и система становится микрогетерогенной. Мицелла лиофильного золя - это ассоциат дифильных молекул, лиофильные группы которых обращены к растворителю, а лиофобные группы соединяются друг с другом, образуя ядро. На рис. 38 схематически изображена мицелла Гартли.

| Рис. 38. Мицелла Гартли |
Такая ориентация дифильных молекул в мицелле обеспечивает минимальное поверхностное натяжение на границе "мицелла - дисперсионная среда". Возникает вопрос - почему образование мицелл происходит самопроизвольно, ведь известно, что образование новой фазы всегда требует затраты энергии. Поскольку поверхностное натяжение на границе с дисперсионной средой минимально, энергия, затрачиваемая на образование мицеллы, невелика. Эта энергия с избытком компенсируется выигрышем энергии за счет введения углеводородных хвостов молекул ПАВ в ядро мицеллы, которое состоит из плотной упаковки углеводородных цепей. Таким образом, мицеллообразование сопровождается уменьшением свободной энергии системы.
При достижении определенной концентрации сферические мицеллы начинают взаимодействовать между собой, что приводит к их деформации. Мицеллы стремятся принять цилиндрическую, дискообразную, палочкообразную, пластинчатую формы. Такие мицеллы называются мицеллами Мак-Бена.
В растворах ПАВ количество вещества в мицеллярной форме может во много раз превышать его количество в молекулярном состоянии. Эти формы находятся в равновесии, состояние которого зависит от концентрации:
 Молекулярный раствор Мицеллы Гартли Молекулярный раствор Мицеллы Гартли
   Дискообразные Цилиндрические Пластинчатые Дискообразные Цилиндрические Пластинчатые
 Мицеллы Мак-Бена
Мицеллы Мак-Бена
  Жидкокристаллическая структура Жидкокристаллическая структура
 Гелеобразная структура Гелеобразная структура
 Твердое кристаллическое ПАВ Твердое кристаллическое ПАВ
|
С мицеллярной структурой ПАВ в водных растворов связано одно из важнейших свойств растворов ПАВ - солюбилизация.
Солюбилизация - это включение в ядро мицеллы ПАВ вещества, нерастворимого или слабо растворимого в дисперсионной среде. В результате солюбилизации в воде растворяются углеводороды (в частности, бензин и керосин), а также жиры.
Солюбилизация связана с проникновением в мицеллы веществ, которые называют солюбилизатами. Мицелла и солюбилизат представляют одно целое. Механизм солюбилизации различной природы солюбилизата можно пояснить при помощи рис. 39.

|
| Рис. 39. Сферические мицеллы и солюбилизация различных веществ а - неполярных; б - с одной полярной группой и гидрофобной частью А; в - содержащих несколько полярных групп Б. |
Явление солюбилизации нашло свое применение в технологии получения нанокатализаторов, т.к. позволяет осуществлять контролируемый синтез наночастиц переходных металлов в ядрах мицелл дифильных блок-сополимеров. Данный подход заключается в направленном использовании ядер мицелл в качестве “нанореакторов” для синтеза коллоидных частиц переходных металлов. Например, в растворах блоксополимера полистирол-поли-4-винилпиридина в толуоле: блоки поли-4-винилпиридина образуют ядро мицеллы, а блоки полистирола - корону (рис. 40).

Рис. 40. Схема формирования наночастиц переходных металлов в блоксополимерных мицеллах: а - быстрое восстановление (NaBH4); б - медленное восстановление (N2H4 ´ H2O)
Соль переходного металла, нерастворимая в толуоле, солюбилизирует в ядро блок-сополимерной мицеллы. Дальнейшая обработка металлсодержащих мицелл химическими восстановителями приводит к образованию наночастиц металлов различного размера и морфологии, стабилизированных в ядрах блоксополимерных мицелл (рис.40). При этом морфология наночастиц различна, в зависимости от скорости формирования зародышей, т.е. от силы восстанавливающего агента.
Необходимо отметить, что в данном примере мицеллы блоксополимера полистирол-поли-4-винилпиридина выполняют не только роль "нанореактора", но и роль стабилизатора наночастиц металлов.
МЕТОДЫОПРЕДЕЛЕНИЯ ПОВЕРХНОСТНОГО НАТЯЖЕНИЯ
Величина σ может быть сравнительно легко и с большой точностью определена для легкоподвижных границ раздела фаз жидкость-газ и жидкость-жидкость.
Существующие методы определения поверхностного натяжения разделяются на три основные группы:
IV. Статические методы:
1) метод капиллярного поднятия;
2) метод лежащей или висящей капли (пузырька).
V. Полустатические методы:
1) метод максимального давления образования пузырька;
2) метод отрыва кольца или рамки;
3) метод отрыва пластинки;
4) метод взвешивания и счета капель
VI. Динамические методы:
1) метод капиллярных волн;
2) метод колеблющихся струй.
Статические методы основаны на изучении практически неподвижных поверхностей, находящихся в равновесии с объёмом и не изменяющихся за время измерения. Динамические методы применяются в основном для изучения существенно неравновесных состояний поверхностных слоев жидкости и скорости установления равновесной структуры их поверхности. Динамические методы основаны на том, что некоторые виды колебаний жидкости сопровождаются периодическим растяжением и сжатием её поверхности. Возвращающая сила либо частично, либо полностью обусловлена поверхностным натяжением, которое, может быть вычислено из периода колебаний.
Для границы твердое тело - газ, твердое тело - твердое тело нет прямых методов определения σ (они не могут самопроизвольно менять свою поверхность). Но косвенные указания говорят о том, что σ твердых тел больше, чем жидких.