1. Основные понятия и законы химической термодинамики
1.1 Основные понятия
Термодинамической системой называют тело или группу тел, находящихся в энергетическом взаимодействии и мысленно или физически отделённых от окружающих тел, которые называются внешней или окружающей средой.
Классификация систем:
1) по возможности тепло- и массообмена: изолированные, закрытые, открытые. Изолированная система не обменивается с окружающей средой ни веществом, ни энергией. Закрытая система обменивается с окружающей средой энергией, но не обменивается веществом. Открытая система обменивается с окружающей средой и веществом и энергией. Понятие изолированной системы используется в физической химии как теоретическое.
2) по внутренней структуре и свойствам: гомогенные и гетерогенные. Гомогенной называется система, внутри которой нет поверхностей, делящих систему на части, различные по свойствам или химическому составу. Примерами гомогенных систем являются водные растворы кислот, оснований, солей; смеси газов; индивидуальные чистые вещества. Гетерогенные системы содержат внутри себя естественные поверхности. Примерами гетерогенных систем являются системы, состоящие из различных по агрегатному состоянию веществ: металл и кислота, газ и твёрдое вещество, две нерастворимые друг в друге жидкости.
Фаза – это гомогенная часть гетерогенной системы, имеющая одинаковый состав, физические и химические свойства, отделённая от других частей системы поверхностью, при переходе через которую свойства системы меняются скачком. Фазы бывают твёрдые, жидкие и газообразные. Гомогенная система всегда состоит из одной фазы, гетерогенная – из нескольких. По числу фаз системы классифицируются на однофазные, двухфазные, трёхфазные и т.д.
|
|
Компонент – всякое вещество, входящее в систему, которое из неё можно выделить и которое может существовать отдельно от системы. По числу компонентов системы классифицируются на однокомпонентные, двухкомпонентные, трёхкомпонентные и т.д.
Свойства системы в физической химии могут быть описаны заданием параметров системы. В качестве параметров чаще всего выступают температура (Т), давление (Р), объём (V), количество вещества (ν) и другие.
Если параметры системы постоянны, говорят, что система находится в состоянии равновесия.
Если параметры системы меняются, то в системе протекает термодинамический процесс. Процесс называют равновесным, если его можно представить как ряд последовательных равновесных состояний системы. В противном случае говорят о неравновесном процессе. Различают обратимые и необратимые процессы. Обратимым называют равновесный процесс, который может в одних и тех же условиях самопроизвольно протекать как в прямом, так и в обратном направлениях. К необратимым процессам относятся неравновесные и несамопроизвольные процессы.
Процессы могут быть: изотермическими (протекают при Т=const), изобарическими (Р=const), изохорическими (V=const), адиабатическими (протекают без теплообмена с окружающей средой). Наибольшее значение в химической термодинамике имеют изобарно-изотермические (Р,Т=const) и изохорно-изотермические (V,Т=const) процессы. Именно в таких условиях протекают все химические реакции.
|
|
1.2 Основные величины
Теплота (Q) – энергия, которая передаётся одной системой другой при их взаимодействии, зависящая только от температур этих систем.
Работа (A) – энергия, передаваемая одной системой другой, зависящая от наличия силового поля или внешнего давления, под действием которого система меняет свой объём. В последнем случае говорят о работе сил расширения.
Правило знаков для теплоты и работы: теплота считается положительной, если она подводится к системе из окружающей среды (поглощённая теплота) и отрицательной в противоположном случае (отданная теплота); работа считается положительной, если она совершается системой над окружающей средой, и отрицательной, если работу совершает окружающая среда над системой.
Внутренняя энергия (U) – запас энергии системы. Включает в себя все виды энергии, связанные со строением системы, и не включает кинетическую и потенциальную энергии системы как целого. Так как абсолютных знаний о строении вещества не существует, абсолютное значение внутренней энергии найти нельзя.
Энтальпия – запас энергии системы в виде теплоты. Связана с внутренней энергией уравнением H = U + PV. Внутренняя энергия, энтальпия, теплота и работа измеряются в Дж/моль. Внутренняя энергия и энтальпия являются, а теплота и работа не являются функцией состояния системы.
Функцией состояния системы называется функция, изменение которой зависит только от начального и конечного состояний системы и не зависит от пути перехода системы из начального в конечное состояние.
Изменения термодинамических функций в химической термодинамике обозначаются по-разному. Если речь идёт о конечном (большом) изменении, то используют греческий символ ∆. Например, ∆Н, ∆U. Бесконечно малые изменения функций, являющихся функциями состояния системы, обозначают латинской буквой d (dU, dH). Если же функция не является функцией состояния системы, то её бесконечно малое изменение обозначается греческой буквой δ (δА, δQ). Изменения функций состояния системы рассчитываются как разность значений данной функции в конечном и исходном состояниях. Например,
|
|
∆Н = Н2 – Н1.
Энтропия (S) – термодинамическая функция, количественно характеризующая степень беспорядка в системе. Является функцией состояния системы, измеряется в Дж/моль∙К.
Энергия Гельмгольца (F) – функция состояния системы, характеризующая протекание химических процессов в изохорно-изотермических условиях.
Энергия Гиббса (G) – функция состояния системы, характеризующая протекание химических процессов в изобарно-изотермических условиях. Энергии Гельмгольца и Гиббса измеряются в Дж/моль.
Соотношение между основными термодинамическими функциями представлено на рис.1.
 |
Н
PV U
PV F TS
G TS
Рисунок 1 - Соотношение между термодинамическими функциями
Теплоёмкость (С) – количество сообщённой системе теплоты, отнесённое к наблюдаемому при этом повышению температуры:
С =  .
.
Различают теплоёмкость при постоянном объёме СV и теплоёмкость при постоянном давлении СP:
СV = 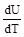 , СP =
, СP = 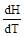 , СP – CV = R,
, СP – CV = R,
где R = 8,314 Дж/моль∙К – универсальная газовая постоянная.
1.3 Законы термодинамики
Законы термодинамики являются эмпирическими, т.е. установлены путём обобщения экспериментальных данных. Первоначально сформулированы для описания работы тепловых машин в середине XIX века. Впоследствии была установлена их универсальность.
Первый закон термодинамики является частным случаем одного из важнейших законов естествознания – закона сохранения и превращения энергии. Применительно к описанию работы тепловых машин он утверждает, что нельзя создать тепловую машину, которая совершает механическую работу без затраты теплоты. Такая тепловая машина получила название вечного двигателя 1-го рода.
Общенаучная формулировка первого закона термодинамики:
Теплота, поглощённая системой, расходуется на изменение внутренней энергии и совершение системой работы:
δQ = dU + δА
Если единственным видом работы является работа сил расширения, то
δQ = dU + PdV
Важнейшим следствием первого закона термодинамики является закон Гесса, позволяющий рассчитывать тепловые эффекты химических реакций.
Второй закон термодинамики определяет условия протекания самопроизвольных процессов. Его первоначальные формулировки касались описания работы тепловых машин. Некоторые из них:
Постулат Клаузиуса: невозможен самопроизвольный переход теплоты от менее нагретого тела к более нагретому телу.
Постулат Оствальда: невозможно создать такую тепловую машину, которая ВСЮ поглощённую теплоту превращает в работу (вечный двигатель второго рода).
Общенаучная формулировка второго закона термодинамики:
Существует функция состояния системы (энтропия), изменение которой следующим образом связано с поглощённой теплотой и температурой системы:
δQ ≤ TdS для самопроизвольных процессов,
δQ = TdS для обратимых процессов,
δQ ≥ TdS для несамопроизвольных процессов.
Второй закон термодинамики позволяет определить направление протекания химических реакций и условия установления химического равновесия.
Третий закон термодинамики описывает протекание процессов при температурах, близких к нулю Кельвина. Он утверждает, что нельзя охладить систему до нуля градусов Кельвина. Абсолютный ноль недостижим. Важнейшим выводом из третьего закона термодинамики является положение о том, что энтропия любого вещества при 0оК равна нулю. Таким образом, в отличие от внутренней энергии и энтальпии значение энтропии вещества при любой температуре – абсолютная энтропия.
1.4 Термодинамические потенциалы
Четыре функции состояния системы U, H, F, G объединены общим названием – термодинамические потенциалы. Для систем, состоящих из одного вещества, количество которого не меняется, можно записать:
dU = TdS – PdV, U = f(S,V),
dH = TdS + VdP, H = f(S,P),
dF = -SdT – PdV, F = f(T,V),
dG = -SdT + VdP, G = f(T,P).
Переменные (S,V,P,T), от которых зависят термодинамические потенциалы, называются естественными переменными. Физический смысл дифференциалов термодинамических потенциалов зависит от того, какие из четырёх переменных постоянны. Если изменяются только две естественные переменные (S,V), а две другие (P,T) остаются постоянными, то изменение внутренней энергии (dU) означает обмен энергией между системой и окружающей средой и в виде теплоты (TdS), и в виде работы (PdV). Изменение энтальпии dH (при постоянных V и T) означает обмен энергией только в виде теплоты. Действительно, первое слагаемое TdS = δQ, а второе слагаемое VdP при V = const означает теплопередачу в изохорическом процессе. Изменение dF при постоянных S и P показывает, что происходит обмен энергией только в виде работы (SdT = δА в равновесном адиабатическом процессе). Изменение dG при постоянных S и V указывает на отсутствие обмена энергией между системой и окружающей средой.
Условия самопроизвольности протекания процессов:
dU ≤ 0 (S,V=const),
dH ≤ 0 (S,P=const),
dF ≤ 0 (T,V=const),
dG ≤ 0 (T,P=const).
В системе могут самопроизвольно протекать только процессы, сопровождающиеся уменьшением термодинамических потенциалов.
Следовательно, все самопроизвольные процессы прекращаются только тогда, когда термодинамические потенциалы достигают минимальных значений и система приходит в состояние равновесия.
Условиями равновесия являются:
dU = 0, d2U>0 (S,V=const),
dH = 0, d2H>0 (S,P=const),
dF = 0, d2F>0 (T,V=const),
dG = 0, d2G>0 (T,P=const).
Наибольшее практическое значение имеют два последних условия, т.к. большинство химических реакций протекают в изохорно-изотермических и изобарно-изотермических условиях.
2. Основы термохимических расчётов
2.1 Основные понятия термохимии
Термохимия – раздел химической термодинамики, изучающий тепловые эффекты химических реакций. В термохимии выделяют два типа реакций. Реакции, протекающие с выделением теплоты, называются экзотермическими, а протекающие с поглощением теплоты – эндотермическими. Среди самопроизвольно протекающих реакций примерно 95% составляют экзотермические реакции. В этом проявляется принцип наименьшей энергии, который говорит о том, что устойчивому состоянию системы отвечает её состояние с минимальной энергией. Поэтому всякая система стремится свою энергию понизить.
Из первого закона термодинамики вытекает, что тепловой эффект химической реакции, протекающей в изохорно-изотермических условиях (в закрытой системе), равен изменению внутренней энергии (QV = ∆U). Если же химическая реакция протекает в изобарно-изотермических условиях (в открытой системе), то eё тепловой эффект равен изменению энтальпии (QP = ∆H). Так как большинство химических реакций протекает в изобарно-изотермических условиях, их тепловой эффект называют энтальпией химической реакции.
Термохимическим уравнением химической реакции называют уравнение химической реакции, в котором указаны её тепловой эффект и агрегатные состояния реагентов и продуктов реакции:
С3Н6О(г)+4О2(г)=3СО2(г)+3Н2О(ж) ∆H=-1817,0 КДж/моль.
Такая запись означает, что в реакции 1 моль газообразного ацетона с 4 моль газообразного кислорода получается 3 моль газообразного углекислого газа и 3 моль жидкой воды. При этом выделяется 1817,0 КДж теплоты. С термохимическими уравнениями можно поступать как с алгебраическими: их можно умножать на какое-либо число, складывать с другими термохимическими уравнениями. Но при этом необходимо и с тепловым эффектом выполнять те же операции.
Так как тепловые эффекты реакций зависят от условий их протекания, то для проведения термохимических расчётов нужны термохимические величины, отнесённые к каким-то одинаковым условиям. В противном случае данные будут несопоставимы. За такие условия принимаются стандартные условия. Если вещество находится при стандартных условиях, его состояние называют стандартным состоянием. За стандартное состояние принимают устойчивое состояние вещества при Т = 298 К и Р = 101325 Па. Поэтому тепловой эффект реакции в стандартных условиях обозначают ∆H298.
2.2 Закон Гесса. Уравнение Кирхгофа
Закон Гесса утверждает:
Тепловой эффект химической реакции зависит только от вида и состояния исходных веществ и продуктов реакции и не зависит от её пути.
Из закона Гесса вытекает ряд следствий:
Тепловой эффект прямой реакции равен по величине и противоположен по знаку тепловому эффекту обратной реакции. Из этого следует, что если прямая реакция экзотермическая, то обратная - эндотермическая.
Если совершаются две реакции, приводящие из двух различных начальных состояний (Н1 и Н2) к одному и тому же конечному состоянию (К), то разность между тепловыми эффектами этих реакций равна тепловому эффекту превращения одного начального состояния в другое.
Если совершаются две реакции, приводящие из одного начального состояния (Н) к двум разным конечным состояниям (К1 и К2), то разность между тепловыми эффектами этих реакций равна тепловому эффекту превращения одного конечного состояния в другое.
∆H1 = -∆H2 ∆H12 = ∆H1 - ∆H2 ∆H12 = ∆H1 - ∆H2
Закон Гесса и его следствия позволяют рассчитывать тепловые эффекты некоторых реакций. Гораздо большее значение для расчётов тепловых эффектов любых реакций имеет правило, вытекающее из закона Гесса.
Для расчёта энтальпий реакций при стандартных условиях ∆H298 необходимо знать энтальпии образования реагирующих веществ и продуктов реакции ∆fHo298. Пусть необходимо рассчитать стандартную энтальпию реакции
n N + m M = d D + g G.
Воспользуемся правилом:
Стандартная энтальпия химической реакции равна разности энтальпий образования продуктов реакции и энтальпий образования исходных веществ с учётом коэффициентов перед веществами в уравнении реакции, т.е.
∆H298=[d∙∆fHo298(D)+g∙∆fHo298(G)] - [n∙∆fHo298(N)+m∙∆fHo298(M)].
Это же правило можно использовать для расчёта стандартных изменений и других функций состояния, например, для расчёта изменения энтропии химической реакции:
∆S298=[d∙So298(D)+g∙So298(G)] - [n∙So298(N)+m∙So298(M)].
В этом случае из таблицы термодинамических величин нужно взять стандартные энтропии веществ So298.
Описанный подход не применим для расчёта изменения функций состояния системы для нестандартных условий, так как отсутствуют необходимые для такого расчёта справочные данные. В этом случае необходимо воспользоваться уравнением Кирхгофа, которое устанавливает зависимость изменения энтальпии или энтропии реакции от температуры:
∆HT = ∆H298 + ∆a∙(T – 298) + ∆b/2∙(T2 – 2982) + ∆c/3∙(T3 – 2983) –∆c’∙(1/T – 1/298),
∆ST = ∆S298 + ∆a∙ln(T/298) + ∆b∙(T – 298) + ∆c/2∙(T2 – 2982) –∆c’/2∙[(1/T2 – 1/2982)].
Здесь ∆a, ∆b, ∆c, ∆c’ – изменения соответствующих коэффициентов в химической реакции. Для расчёта этих величин необходимо в справочнике найти коэффициенты a, b, c, c’ и рассчитать изменения по общепринятой в термодинамике методике. Например,
∆a=[d∙a(D)+g∙a(G)] - [n∙a(N)+m∙a(M)].
Для расчёта изменения энергии Гиббса ∆G химической реакции следует воспользоваться формулой
∆GT = ∆HT – T ∙ ∆ST,
где Т – любая (стандартная или нестандартная) температура. При расчётах по последней формуле необходимо использовать значения ∆H и ∆S, соответствующие этой температуре.
2.3 Расчёты изменения термодинамических функций химических реакций
Проведём расчёт изменений энтальпии, энтропии и энергии Гиббса химической реакции
4 СО(г) + 2 SO2(г) = S2(г) + 4 CO2(г)
для стандартной (298К) и нестандартной (500К) температур. Перед началом расчётов необходимо ещё раз убедиться в том, что реакция уравнена.
Рассчитаем сначала ∆H298, ∆S298 и ∆G298.
∆H298=[∆fHo298(S2)+4∙∆fHo298(CO2)]- [4∙∆fHo298(CO)+2∙∆fHo298(SO2)]=[128,37+4∙ (-393,51)]–[4∙ (-110,53) +2∙ (-296,90)]= - 409,75 КДж.
∆S298=[ So298(S2)+4∙So298(CO2)] - [4∙So298(CO)+2∙So298(SO2)] =(228,03+4∙213,66)–(4∙197,55+2∙248,07) = - 203,67 Дж/К.
∆G298=∆H298–298∙∆S298=-409750 – 298∙(-203,67)= - 349056 Дж.
Расчёт показывает, что изучаемая экзотермическая реакция (знак энтальпии) при стандартных условиях может протекать самопроизвольно (знак энергии Гиббса).
Для расчёта нестандартных величин по уравнениям Кирхгофа требуется рассчитать ∆a, ∆b, ∆c, ∆c’. Для удобства и компактности расчётов составим таблицу 1.
Таблица 1 - Расчёт ∆a, ∆b, ∆c, ∆c’
| Номер строки | Вещество | Cp = f(T), Дж/моль∙К | |||
| a | b∙103 | c’∙10-5 | c∙106 | ||
| S2 | 36,11 | 1,09 | -3,51 | ||
| CO2 | 44,14 | 9,04 | -8,54 | ||
| 4 CO2 | 176,56 | 36,16 | -34,16 | ||
| ∑кон a,b,c,c’ | 212,67 | 37,25 | -37,67 | ||
| CO | 28,41 | 4,10 | -0,46 | ||
| 4 CO | 113,64 | 16,40 | -1,84 | ||
| SO2 | 46,19 | 7,87 | -7,70 | ||
| 2 SO2 | 92,38 | 15,74 | -15,40 | ||
| ∑исх a,b,c,c’ | 206,02 | 32,14 | -17,24 | ||
| ∆a,∆b,∆c,∆c’ | 6,65 | 5,11 | -20,43 |
Строки 1, 2, 5 и 7 содержат справочные значения всех коэффициентов. Данные строк 3, 6 и 8 являются результатом умножения чисел в строках 2, 5 и 7 на соответствующий множитель (коэффициент перед данным веществом в уравнении реакции). Цифры в 10-й строке – результат вычитания данных 9-й строки из данных 4-й строки. Коэффициент а для всех веществ имеет истинное значение. Остальные коэффициенты либо увеличены, либо уменьшены в 10n раз. Это сделано для компактности таблицы 1 (общепринятый способ представления табличных данных). Истинные значения коэффициентов b, c, c’ равны значащим цифрам из таблицы 1, умноженным на 10-n, т.е. знак показателя степени множителя следует изменить на противоположный. Коэффициент с для всех веществ изучаемой реакции равен нулю. Рассчитаем ∆H500.
∆H500 = ∆H298 + ∆a ∙ (500–298) + ∆b/2 ∙ (5002 – 2982) + ∆c/3 ∙ (5003 - 2983) – ∆c’ ∙ (1/500 – 1/298) = - 409750 + 6,65 ∙ (500 – 298) + 5,11 ∙ 103/2 ∙ (5002 - 2982) – (-20,43∙105)∙(1/500 –1/298) =-409750+1343+412–2770= -410765 Дж.
Видно, что рассчитанное значение незначительно отличается от стандартного.
Рассчитаем ∆S500.
∆S500 = ∆S298 + ∆a∙ln(500/298) + ∆b∙(500 – 298) + ∆c/2∙(5002 – 2982) – ∆c’/2∙[(1/5002 – 1/2982)] = - 203,67 + 6,65∙ln(500/298)+ 5,11∙10-3 (500 – 298) - (- 20,43∙105/2) ∙ (1/5002 – 1/2982) = -203,67 + 3,44 + 1,03 – 7,42 = -206,62 Дж/моль∙К.
Найдём изменение энергии Гиббса ∆G500.
∆G500 = ∆H500 – 500 ∙ ∆S500 = -410765-500∙ (-206,62) = -307455 Дж.
Изучаемая реакция может протекать самопроизвольно и при 500 К.
3. Химическое равновесие
3.1 Константа химического равновесия
Все химические реакции в той или иной мере обратимы, т.е. не идут до конца, до полного превращения исходных веществ в продукты. В реакционной смеси всегда происходит как прямая, так и обратная реакции. По мере расходования исходных веществ скорость прямой реакции снижается, по мере накопления продуктов возрастает скорость обратной реакции. Когда эти скорости сравниваются, устанавливается динамическое равновесие: не происходит ни накопления, ни расходования исходных веществ и продуктов. Суммарная скорость прямой и обратной реакций будет равна нулю. Такое состояние системы называется состоянием химического равновесия.
С термодинамической точки зрения состояние равновесия характеризуется равенством нулю изменения энергии Гиббса реакции ∆G = 0. При этом подразумевается, что энергия Гиббса является функцией не только температуры и давления, но и количеств всех веществ, входящих в систему.
Количественной характеристикой химического равновесия служит константа химического равновесия. В зависимости от того, в какой системе протекает химическая реакция, константа равновесия может выражаться по-разному.
Пусть в системе протекает обратимая химическая реакция между газообразными веществами N и M и образуются газообразные вещества D и G.
n N + m M ↔ d D + g G.
Константа химического равновесия в этом случае может быть найдена как
КР =  .
.
В этой формуле все Рi – парциальные давления пара всех компонентов равновесной газовой смеси. Размерность КР в соответствии с формулой будет [Паd+g-n-m]. Если какое-то вещество не газообразное, например, вещество М – твёрдое, то, учитывая, что давление пара над твёрдым веществом постоянно, выражение для константы равновесия примет вид
КР =  .
.
В соответствии с этим изменится и размерность константы скорости - [Паd+g-n].
Если рассматриваемая реакция протекает в растворе, то константу химического равновесия выражают через равновесные молярные концентрации
КС = 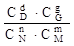 .
.
Соответственно изменится и размерность [(моль/л)d+g-n-m].
Обе эти константы равновесия связаны уравнением
КР = КС∙ (RT)d+g-n-m.
Из этого уравнения видно, что если реакция протекает без изменения числа газообразных молекул, то КР = КС.
Численное значение константы химического равновесия характеризует глубину протекания прямой и обратной реакций. Так, если К>>1, это означает, что преимущественно протекает прямая реакция. Если же К<<1, то при данных условиях глубже протекает обратная реакция.
Константа химического равновесия связана с термодинамическими потенциалами и может быть рассчитана через их значения:
ln KP = -  ,
,
ln KC = -  ,
,
lnKP = - 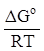 ,
,
lnKC = -  .
.
Рассчитаем константу химического равновесия реакции 4 СО(г) + 2 SO2(г) = S2(г) + 4 CO2(г) при стандартной и нестандартной (500 К) температурах. Воспользуемся для этого уравнением, связывающим КР и ∆G.
ln KP(298) = - (- 349056/8,314∙298). Отсюда КР = 1,535∙1061 Па-1.
ln KP(500) = - (- 307455/8,314∙500). Отсюда КР = 1,321∙1032 Па-1.
3.2 Смещение химического равновесия
Достигнув состояния химического равновесия, система будет находиться в нём до тех пор, пока не будут изменены внешние условия. Это приведёт к изменению параметров системы, т.е. к сдвигу химического равновесия в сторону одной из реакций. Для качественного определения направления смещения равновесия в химической реакции служит принцип Ле-Шателье – Брауна:
Если на систему, находящуюся в равновесии, оказать внешнее воздействие, т.е. изменить условия, при которых система находилась в равновесии, то в системе с большей скоростью начнут протекать процессы, УМЕНЬШАЮЩИЕ оказанное воздействие.
На состояние химического равновесия наибольшее влияние оказывают концентрация, давление, температура.
Как видно из выражения для константы скорости реакции, увеличение концентраций исходных веществ N и M приводит к возрастанию скорости прямой реакции. Говорят, что равновесие сдвинулось в сторону прямой реакции. Наоборот, увеличение концентраций продуктов смещает равновесие в сторону протекания обратной реакции.
При изменении общего давления в равновесной смеси парциальные давления всех участников реакции изменяются в одинаковое число раз. Если в реакции число моль газов не изменяется, как, например, в реакции H2 + Cl2 ↔ 2 HCl, то состав смеси остаётся равновесным и равновесие не смещается. Если же число моль газов в реакции изменяется, то состав смеси газов в результате изменения давления станет неравновесным и одна из реакций начнёт протекать с большей скоростью. Направление смещения равновесия в этом случае зависит от того, увеличилось или уменьшилось число моль газов.
Рассмотрим, к примеру, реакцию
N2 + 3 H2 ↔ 2 NH3
Все участники этой реакции – газы. Пусть в равновесной смеси увеличили общее давление (сжали смесь). Равновесие нарушится, в системе должны начаться процессы, которые приведут к уменьшению давления. Но давление пропорционально числу ударов молекул о стенки, т.е. числу молекул. Из уравнения реакции видно, что в результате протекания прямой реакции число молекул газов уменьшается с 4 моль до 2 моль, а в результате обратной соответственно увеличивается. Следовательно, уменьшение общего давления произойдёт, если равновесие сместится в направлении протекания прямой реакции. При уменьшении общего давления в этой системе равновесие сместится в направлении протекания обратной реакции, приводящей к увеличению числа молекул газов, т.е. к увеличению давления.
В общем случае при повышении общего давления равновесие смещается в сторону реакции, приводящей к уменьшению числа молекул газообразных веществ, а при уменьшении давления – в сторону реакции, в которой увеличивается число молекул газов.
Для определения направления смещения равновесия при изменении температуры системы необходимо знать тепловой эффект реакции, т.е. экзотермическая данная реакция или эндотермическая. При этом нужно помнить, что при протекании экзотермической реакции теплота выделяется и температура повышается. При протекании эндотермической реакции температура падает за счёт поглощения теплоты. Следовательно, при повышении температуры равновесие всегда смещается в сторону эндотермической реакции, а при понижении – в сторону экзотермической реакции. Например, в системе, где протекает обратимая реакция
N2 + 3 H2 ↔ 2 NH3, ∆H298 = - 92,4 КДж/моль.
При повышении температуры равновесие сместится в сторону обратной (эндотермической) реакции, а при понижении температуры – в сторону прямой реакции, которая экзотермическая.