Генетические причины умственной отсталости
Умственная отсталость является одной из основных причин обращения в генетическую консультацию. Генетическими причинами обусловлено до половины случаев тяжёлой умственной недостаточности. Основные типы генетических нарушений, ведущих к умственной недостаточности, включают:
· Хромосомные аномалии, нарушающие дозовый баланс генов, такие как анеуплоидия, делеции, дупликации. Примерами могут служить трисомия хромосомы 21 (синдром Дауна), частичная делеция короткого плеча хромосомы 4, микроделеция участка хромосомы 7q11.23 (синдром Вильямса) и др.
· Дерегуляция импринтинга вследствие делеций, однородительской дисомии хромосом или участков хромосом. Этим механизмом обусловлены такие заболевания, как синдром Ангельмана и синдром Прадера-Вилли.
· Дисфункция отдельных генов. Число генов, мутации в которых вызывают ту или иную степень умственной отсталости, превышает 1000. В их число входят, например, ген NLGN4, находящийся на хромосоме Х, мутации в котором обнаружены у некоторых пациентов, страдающих аутизмом; ген FMR1, сцепленный с хромосомой Х, дерегуляция экспрессии которого вызывает синдром хрупкой Х-хромосомы; ген MECP2, также находящийся на хромосоме Х, мутации в котором вызывают синдром Ретта у девочек[13].
Наследственные формы олигофрении. Хромосомные нарушения
Все формы олигофрении отчетливо распадаются на несколько вариантов в зависимости от этиологии и патогенеза расстройства. Г.Е.Сухарева (1965) разграничила следующие группы олигофрении: 1) олигофрении эндогенной природы, связанные с повреждением наследственного аппарата; 2) бластопатии; 3) эмбриопатии; 4) ранние и поздние фетопатии; 5) олигофрении, возникающие вследствие родовой травмы, а также различной патологии в первые три года жизни ребенка. Наиболее часто олигофрении (до 90%) связаны с генетическими факторами, но это не общее мнение.
Наследственные формы олигофрении
Синдромы олигофрении c хромосомными аберрациями, мутациями генов
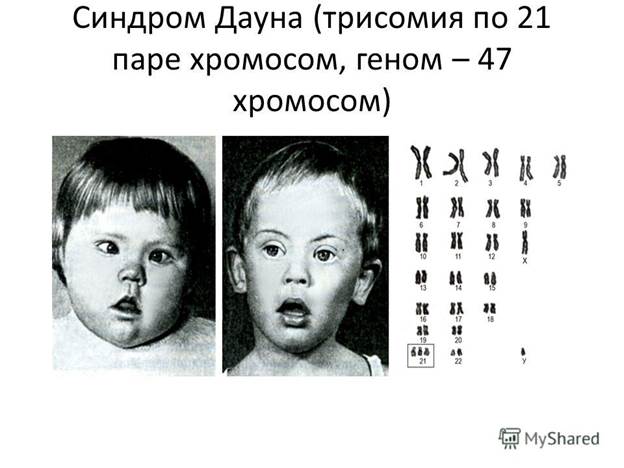
1. Болезнь Дауна (1886). Встречается у одного новорожденного на 700. Умственная отсталость у 75% пациентов достигает степени имбецильности, у 20% — идиотии, у 5% — дебильности. У большинства пациентов выявляется генетический дефект в виде трисомии 21. (Трисомия означает наличие в хромосомном наборе дополнительной, третьей хромосомы, в то время, когда норма предписывает лишь пару. Точные причины трисомии по 21 хромосоме не установлены, однако механизм её формирования заключается в том, что при делении клеток не происходит расхождения хромосом (чаще женских) и образуется клетка с 24 хромосомами. В процессе слияния яйцеклетки и сперматозоида, клетка с 24 хромосомами сливается с нормальной клеткой с 23 хромосомами. В итоге образуется зигота с 47 хромосомами (23 пары + одна хромосома), вместо 46.) Генетический дефект может иметь мозаичный характер: часть клеток тела являются нормальными, часть — трисомичными. Встречаются транслокации, когда 21-я хромосома сцепляется с другой, чаще с 15-й аутосомой. Даун отмечал, что частота этого заболевания составляет 10% среди всех больных с умственной отсталостью. Матери средних лет (более 32) рискуют родить больного ребенка в семь раз чаще. Частота рождения пациента с синдромом Дауна возрастает и при очень ранних родах. Если же имеет место транслокация хромосом, риск иметь больного ребенка увеличивается до 1:3.
Диагноз болезни может быть установлен посредством амниоцентеза (пункции амниотической оболочки с целью получения околоплодных вод для последующего лабораторного исследования...)между 14-й и 16-й неделями беременности с последующим изучением клеток околоплодных вод с целью своевременного прерывания беременности. Умственное развитие больных детей как будто бы происходит нормально с рождения до шести месяцев. Затем начинают выявляться признаки задержки умственного развития. Согласно многим сообщениям, дети с синдромом Дауна большей частью являются спокойными, добродушными, общительными, способными привязываться, что облегчает их адаптацию в домашних условиях. Теория врожденного преступника
Ломброзо здесь явно опровергается. В подростковом возрасте, особенно если дети живут в интернатах, могут обнаруживаться эмоциональные расстройства, а в редких случаях — и психотические эпизоды с обманами восприятия, возбуждением. Диагноз затруднителен у новорожденных младенцев. Наиболее серьезными признаками расстройства являются общая гипотония, косые пальпебральные щели, избыток кожи на шее, маленький уплощенный череп, высокие скулы и высунутый язык. Руки широкие и толстые, с одной поперечной ладонной складкой, и короткие пальцы, закругленные внутрь. Рефлекс Моро ослаблен или отсутствует (в норме на похлопывание по животу, грудной клетке, ягодицам, дуновение в лицо руки отводятся, вытягиваются по оси тела, при этом находятся почти перпендикулярно к туловищу, а затем вновь возвращаются к срединной линии тела).
При синдроме Дауна описано более 100 признаков и стигм, но все вместе они редко встречаются у одного пациента. Часто встречаются пороки строения внутренних органов, эндокринные нарушения, недоразвитие половых желез и вторичных половых признаков, ломкость волос, ногтей, очаги облысения. Повышена частота лейкозов. Выявляются также слабость конвергенции, косоглазие, нарушение вестибулярного аппарата, признаки вегетодистонии. У 9–10% больных наблюдается судорожный синдром. Поздно происходит половое созревание, рано наступает инволюция (в 30–40 лет). Большинство пациентов не доживает до 40 лет; старение напоминает картину болезни Альцгеймера. Типичное строение лица дало другое название болезни — «монголоидная идиотия»: микробрахицефалия, близкое расположение и косой разрез глаз с кожной складкой во внутреннем углу (эпикант), широкая и запавшая переносица, полуоткрытый рот, неправильная форма ушей, готическое небо. Лечения болезни не существует.
2. Синдром Аарского. Наблюдаются гипертелоризм (повышенное расстояние между парными органами, расположенными симметрично; чаще всего между глазами), широкий мостик носа, антевертированные (развернутые вперед) ноздри, длинный губной желобок. Короткие руки и ноги, легкая перепончатость между пальцами, скротальное «покрывало» над половым членом, маленький рост. Может быть умственное недоразвитие. Тип генетической трансмиссии: Х-примыкающий полудоминантный.
3. Синдром Апера (акроцефалосиндактилия), 1906. Характерен башенный череп с резким уплощением затылка и нависающими лобными отделами. Уродливое строение лицевой части черепа: лунообразное лицо, деформированные ушные раковины, экзофтальм, гипертелоризм, седловидное переносье. Краниостеноз, непостоянная среднефациальная гипоплазия, иногда — незаращение мягкого и твердого неба, недоразвитие верхней и прогнатизм нижней челюсти. Синдактилия на руках и ногах с дистальным слиянием мягких тканей и вовлечением костных структур, иногда один общий коготь для нескольких пальцев. Атрофия зрительных нервов. Расширение дистальных отделов пальцев рук и ног, аномалии строения внутренних органов. Нарушена глазодвигательная иннервация. Умственная отсталость варьирует от легкой дебильности до идиотии. Одновременно выявляются симптомы психоорганического синдрома (вспыльчивость, импульсивность, склонность к агрессии). Иногда интеллект не нарушен. Тип генетической трансмиссии аутосомно-доминантный.
4. Синдром Сотоса (церебральный гигантизм) (1964). Типично сочетание олигофрении и ожирения. Умственная отсталость нерезко выражена (более выражена в раннем возрасте). Пациенты могут быть возбуждены, агрессивны, злобны. В детском возрасте отмечаются чрезвычайно быстрый рост тела, большая голова, выступающий лоб, узкая нижняя челюсть. Большие руки и ноги, акромегалия, гипертелоризм, антимонголоидный разрез глаз, прогнатизм. Бывают долихоцефалия, гиперемия и одутловатость лица, макроглоссия, сколиоз. На Р-грамме — ускорение костного возраста. Повышена склонность к простудным болезням. Пациенты моторно неловки, неуклюжи, угловаты, дискоординированы. Рост тела не всегда бывает чрезмерным. Тип генетической трансмиссии неизвестен. В патогенезе важное значение придается поражению гипоталамуса.
5. Синдром Коккейна-Нейла (микроцефалический, кахектический тип нанизма). В течение первого года жизни дети развиваются обычно нормально, на втором-третьем году наступает регрессия и выявляются соответствующие симптомы. Основными из них являются микроцефалия, кахексия, отставание умственного развития, очень узкое лицо с близко посаженными глазами, энофтальм и тонкий клювовидный нос. Выявляются, кроме того, пигментное перерождение сетчатки, иногда — атрофия зрительного нерва и катаракта. Наблюдаются признаки прогерии («раннего старения»): раннее поседение, дряблость кожи, снижение слуха и зрения, нарушения светочувствительности. Обычными симптомами являются также недоразвитие мышц, подкожно-жирового слоя, ранняя эпифизарная оссификация, ведущая к срастанию эпифизов и тем самым к нарушениям подвижности в локтевом и коленном суставах. Вторичные половые признаки недоразвиты. Встречаются спленомегалия, удлиненные и увеличенные руки и ноги, гипотрихоз. Тип генетической трансмиссии, предположительно, аутосомно-рецессивный. Лечение симптоматическое.
6. Синдром Кохена. Типичны гипоплазия нижней челюсти с выдающимися резцами, узкие руки и ноги. Наблюдаются также мышечная гипотония и ожирение. Выражена умственная отсталость. Рост тела может быть как увеличен, так и уменьшен. Тип генетической трансмиссии, предположительно, аутосомно-рецессивный. Специфического лечения не существует.
7. Синдром Корнелия де Ланге, или амстердамская карликовость, описан в 1933 г. Карликовость (нанизм, микросомия) отмечается при росте до 130–140 см в зрелом возрасте. Пациентам с упомянутым синдромом свойственны синофриз (сросшиеся брови), микроцефалия с недоразвитием лобных и других отделов мозга, тонкая и повернутая книзу нижняя губа, длинный губной желобок, а также вывернутые вперед ноздри. Наблюдается масса прочих краниофациальных дисгенезий: длинные густые загнутые ресницы, короткий нос, сдавленная переносица, увеличенное расстояние между основанием носа и верхней губой.
Руки и ноги маленькие, неправильной формы, большой палец расположен проксимально, нередки синдактилии, нехватка пальцев. Очень часты пороки внутренних органов, особенно почек, у подавляющего большинства пациентов выявляется имбецильность или глубокая дебильность; в части случаев дело ограничивается пограничной умственной отсталостью. У четверти больных встречаются судорожные припадки. Есть сведения о том, что эти пациенты склонны к аутоагрессии, стереотипному манежному бегу, вращению вокруг оси тела, стереотипным движениям руками. О наличии психотических расстройств не сообщается. Тип генетической трансмиссии не установлен. У части пациентов выявлены хромосомные аберрации, в некоторых случаях прослеживается аутосомно-рецессивное наследование. Носители аномальных аллелей гена могут обнаруживать черты внешнего сходства с пациентами и признаки легкой умственной задержки.
8. Синдром Лежена, или «болезнь кошачьего крика». Типичны микроцефалия, круглое лицо, гипертелоризм, эпикантальные складки или скошенные глазные щели. Отмечаются короткие пястные или плюсневые кости, на руке — четыре пальца. В младенчестве дети издают крики, подобные кошачьим, из-за недоразвития голосовых щелей. Тело уменьшено в размерах, выражена умственная отсталость. Тип генетической трансмиссии не установлен. Специфического лечения и эффективной профилактики не существует.
9. Синдром Крузона (Круазо), или черепно-лицевой дизостоз, дискефалия, наследственная черепно-лицевая дисплазия, краниофациальный дизостоз. Описан L.E.O.Crouzon в 1912 г. Умственная отсталость разной степени сочетается с пороками развития костей (дизостеозом). Характерны деформация черепа, гипертелоризм, экзофтальм, неполный птоз (проптоз), дисплазии лица, в частности гипоплазия верхней челюсти. Наблюдаются выраженная и нарастающая гидроцефалия, застой и атрофия дисков зрительных нервов. Прогрессирует снижение зрения, наблюдается косоглазие, в патогенезе, считается, существенную роль играют нарушения кровообращения головы, возникающие на ранних этапах эмбрионального развития. Тип генетической трансмиссии аутосомно-доминантный, а также аутосомно-рецессивный. При лечении могут быть полезны консервативные и оперативные мероприятия, имеющие целью снижение интракраниальной гипертензии.
10. Синдром Дубовица. Дети очень маленького роста. Маленькое лицо, птоз, латеральное смещение нижнего внутреннего угла глаза. Широкая спинка носа. Редкие волосы. Микроцефалия. Наблюдается детская экзема. Умственная отсталость разной степени, иногда весьма незначительной. Тип генетической трансмиссии, предположительно, аутосомнорецессивный. Лечения не существует.
11. Синдром Гольденхара. Наблюдаются гипоплазия скуловой кости (молярная гипоплазия), макростомия (увеличенная ротовая щель), микрогнатия. Кроме того, описаны эпибульбарный дермоид или липодермоид, а также оттопыренные ушные раковины с периаурикулярными полипами. Выявляются позвоночные аномалии. Сведения об умственной отсталости пациентов отсутствуют. Тип генетической трансмиссии не установлен.
12. Синдром диспигментоза. Выявляется деформация ушных раковин, могут быть дефекты зубов и пятнистая плешивость. Главным внешним признаком является кожная пигментация «в крапинку», в виде паутины или в виде «цветков». Умственная отсталость варьирует в широких пределах, от значительной до близкой к норме. Рост тела не нарушен. Тип генетической трансмиссии, предположительно, доминантный Х-примыкающий. Предполагается, что эта патология для особей мужского пола летальна.
13. Синдром Лоренса-Муна-Бидля-Барде (1922), или синдром окулодиэнцефальный. Основные признаки: пигментный ретинит, адипозогенитальная дистрофия и умственная отсталость. Могут быть полидактилия, синдактилия, деформации черепа и скелета, атрезия заднего прохода. Наблюдаются, кроме того, ожирение, гипогенитализм и эпилептические припадки. Степень умственной отсталости варьирует в широких пределах: от значительной до пограничной или даже вплотную приближающейся к норме. Случаев высокого интеллекта не зафиксировано. Рост пациентов может быть увеличенным, уменьшенным, не отличающимся от нормального. Тип генетической трансмиссии аутосомно-рецессивный, носительство гена заметным образом себя не обнаруживает. Распознать болезнь при рождении ребенка можно по характерным дисгенезиям. Отчетливые признаки болезни формируются в младенческом возрасте, в немалой степени этому способствуют менингиты, энцефалиты, ЧМТ. Специфических методов лечения не существует, нет и надежных методов профилактики.
14. Линейный невус. Внешних признаков расстройства немного: это главным образом сальная родинка (невус) на лице и/или на шее. Могут быть эпилептические припадки. Всегда бывает умственная отсталость. Рост может быть как увеличенным, так и уменьшенным. Тип генетической трансмиссии не установлен.
15. Синдром Лоу (1952), или окулоцереброренальный синдром. Болеют только мальчики, заболевание передается только по материнской линии. К числу наиболее характерных признаков болезни относятся поражения глаз (двусторонняя глаукома, катаракта, гидрофтальм). Выраженная умственная отсталость обычно сочетается с нанизмом при пропорциональном сложении тела. Отмечаются крипторхизм, а также недостаточное окостенение скелета. Поражены почки. Дисфункция канальцев обнаруживается альбуминурией и повышенным выделением с мочой органических кислот (гипераминоацидурия). Типична мышечная гипотония. Рост пациентов может быть увеличенным. Выражена во всех случаях умственная отсталость. Патологоанатомически выявляются гипоплазия лобных долей, гидроцефалия, неполная дифференцировка коркового вещества почек. Патология обнаруживается сразу после рождения. Обычно дети погибают в возрасте до 10 лет от присоединения интеркуррентной инфекции (стафилококк) с септикопиемией или от недостаточности почек. Тип генетической трансмиссии Х-примыкающий рецессивный.
16. Синдром Мебиуса, или врожденная лицевая диплегия. Наблюдается паралич глаз, отчего лицо пациента кажется лишенным всякого выражения. Может быть косолапость. Отмечается синдактилия пальцев рук и/или ног. Умственная отсталость выражена не во всех случаях. Рост тела может быть как увеличенным, так и уменьшенным. Тип генетической трансмиссии не установлен.
17. Нейрофиброматоз Реклингаузена (1882). Важный признак болезни у детей — множественные пятна кофейного цвета (до нескольких сантиметров в диаметре), чаще на груди, спине, животе. Они могут быть уже при рождении, но чаще появляются до 10 лет, увеличиваясь в числе и размерах. Патогномонично высыпание мелких кофейных пятен под мышками. Бывают и другие изменения кожи: сосудистые пятна, очаги депигментации, гипертрихоз, очаговое поседение волос, мягкие и светлые опухоли, которые при надавливании проваливаются в кожу. Под кожей по ходу нервов образуются округлые, размером 1–2 см, не спаянные с кожей «плексиформные невриномы». Случается, что их бывает не более 1–2, но обычно намного больше. Невриномы в области черепных нервов нередко нарушают функцию последних (снижение зрения, слуха, боли и др.). Выявляются различные дисплазии: гипертелоризм, деформация черепа, крупная голова и грубое лицо с признаками акромегалоидности, крупные кисти и стопы, короткая шея, вдавленная грудина (куриная грудь). У мальчиков постарше отмечается евнухоидность (высокий таз, длинные ноги, гипогенитализм).
Часты другие пороки развития: вывих тазобедренного сустава, псевдоартрозы, пороки сердца, весьма нередки внутренние опухоли, которые позже подвергаются малигнизации. При локализации опухоли внутри черепа развивается соответствующая симптоматика: афазия, слепота, припадки, деменция, атаксия и др. У половины больных с периферическими опухолями наблюдаются умственная отсталость, чаще неглубокая, отклонения моторики и речи. Частой бывает гидроцефалия, иногда с выраженным беспокойством ребенка. Выраженный умственный дефект иногда сочетается с расторможенностью, резкими аффективными вспышками. У детей школьного возраста часто наблюдают вялость, астению, склонность к ипохондрии, колебания настроения, навязчивости, страхи, нарушения сна. Для периферической формы болезни усиление умственной отсталости нехарактерно. Тип генетической трансмиссии аутосомный доминантный с генами на хромосомах 17 и 22 (в последнем случае заболевание протекает тяжелее).
18. Синдром Улльриха-Нунан (синдром Нунан, синдром Бонневи-Улльриха). Описан в 1930 г. (Улльрих). По своим проявлениям сходен с синдромом Тернера: шейная крыловидная складка или короткая шея, низкий рост, гипертелоризм, эпикант, антимонголоидный разрез глаз, деформированные и низко расположенные уши, узкая верхняя и уменьшенная нижняя челюсти, гонадальный дисгенез, крипторхизм. Наблюдаются аномалии внутренних органов, характерен стеноз легочной артерии. Грудная клетка «образует полость», локти вывернуты наружу. Умственная отсталость чаще всего невысокая, редко бывает глубокой, встречаются и лица с высоким интеллектом. Тип генетической трансмиссии аутосомно-доминантный, связанный с 12-й хромосомой. В случаях высокого сходства с проявлениями синдрома Тернера требуется цитогенетическое исследование. Специфического лечения не существует.
19. Синдром Прадера-Вилли (1956). Типично сочетание олигофрении и церебрального ожирения. Обычно наблюдается выраженная умственная отсталость, иногда с аспонтанностью, на фоне которой эпизодически возникают вспышки агрессии. Обычны малый рост, маленькие руки и ноги, гипогенитализм. Мышечная гипотония, особенно в младенчестве, анорексия, сменяющаяся позднее булимией. Могут быть сердечные аномалии. Диспластические черты: долихоцефалия, деформированные и низко расположенные ушные раковины, мягкий ушной хрящ, миндалевидные глазные щели с их скошенностью по направлению вверх, вертикально, эпикант, гипертелоризм, страбизм, высокое небо, подковообразная форма рта с короткой верхней губой, неправильный рост зубов. У мальчиков — крипторхизм, у девочек — недоразвитие больших и малых половых губ. В пубертатном периоде нередко присоединяется диабет. Тип генетической трансмиссии не установлен. В патогенезе заболевания большое значение придают поражению гипоталамуса. Специфического лечения не существует.
20. Синдром Мартина-Белл (1943). Среди мальчиков с умственной отсталостью составляет 6–10%. Детям свойственна своеобразная манера речи: ускоренная по темпу и с выраженными персеверациями — это, как правило, быстрое повторение целых фраз или их окончаний. Иногда бывают заикание, легкая дизартрия. Умственная недостаточность варьирует от имбецильности до пограничной отсталости. Часто бывает синдром гиперактивности, несколько повышена аффективная возбудимость. У более сохранных пациентов находят боязливость, тормозимость. Многие дети эмоционально вполне адекватны, способны формировать привязанности. Приблизительно у трети пациентов выявляются шизофреноподобные симптомы: аутизм, стереотипные и вычурные движения, повороты вокруг оси своего тела.
Выявляются различные диспластические признаки: большая голова с высоким и широким лбом, большие оттопыренные уши, удлиненное лицо с увеличенным подбородком и несколько уплощенной средней частью. Нос часто бывает клювовидным, но с округлым кончиком и широким основанием. Кисти и стопы увеличены, дистальные фаланги пальцев расширены. Радужка нередко бывает симметрично светлой. Кожа гиперпластична, легко растягивается, повышена разгибаемость суставов. У детей постарше масса тела избыточна. У подростков и больных постарше находят макроорхизм с обычной эндокринной функцией. Встречаются и неврологические симптомы: мышечная гипотония, дискоординация движений, оживление сухожильных рефлексов, тики, атетоидные движения, у 8–10% пациентов — эпилептический синдром. Тип генетической трансмиссии рецессивный, связанный с ломкой (фрагильной) X хромосомой. Отмечают высокую пенетрантность аномального гена: у трети женщин — носительниц этого гена выявляется когнитивный дефицит. В нисходящих поколениях заболевание утяжеляется. Современные знания позволяют выявлять женщин-гетерозигот. Предполагают, что в патогенезе болезни играет роль дефицит фолиевой кислоты. Лечение ею смягчает шизофреноподобную симптоматику и гиперактивность, но на умственное развитие не влияет.
21. Синдром Робина (1929). Характерной является такая триада признаков: глоссоптоз в сочетании с другими аномалиями полости рта, что может повлечь тяжелые нарушения дыхания; микроцефалия; умственное недоразвитие. Могут быть волчья пасть, пороки сердца. Тип генетической трансмиссии аутосомно-рецессивный.
22. Синдром Рубелла. Наблюдаются катаракта, пигментация сетчатки, деформация глаза. Отмечаются, кроме того, сенсонейральная глухота, открытый артериальный проток. Умственная ретардация варьирует от выраженной до весьма умеренной. Тип генетической трансмиссии не установлен.
23. Синдром Рубинстайна-Тейби (1963). Умственная отсталость и задержка роста сочетаются с характерными особенностями строения лица и тела. В числе последних короткий и широкий первый палец на кисти и стопе, своеобразное лицо с длинным загнутым носом, антимонголоидный разрез глаз, гипертелоризм, недоразвитие верхней челюсти, низкий рост волос на лбу, иногда спускающийся углом к центру, брахицефалия. Бывают расширение концевых фаланг других пальцев, синдактилия и полидактилия стоп, косолапость, врожденный вывих бедра, повышенная разгибаемость суставов. Иммунодефицит. Масса тела снижается после рождения. Часты пороки внутренних органов. Весьма типичны нарушения глаз: катаракта, колобомы, аномалии рефракции, глаукома, атрофия зрительных нервов, косоглазие, заражение слезно-носового канала. У четверти больных бывают эпилептические припадки. Умственная ретардация обычно глубокая, но встречаются и пограничная отсталость, и даже варианты нормы. Некоторые пациенты проявляют агрессию, аутоагрессию, склонны к аффективным вспышкам. Тип генетической трансмиссии в некоторых случаях связан с микроделецией хромосомы 16. Специфического лечения не существует.
24. Умственная ретардация с врожденным гипертрихозом и речевым недоразвитием. Описана Г.С.Мариничевой с сотрудниками (1976). Умственная отсталость варьирует в пределах от идиотии, имбецильности до задержки в пограничной степени. Нарушения речи разнообразны, чаще это дизартрия. Нередко речевая активность снижена до моторной алалии, часто ослаблена фонация. Гипертрихоз более представлен на спине, разгибательных поверхностях конечностей, лице. На лице, кроме того, выявляются некоторые дисгенезии: широкий нос, антимонголоидный разрез глаз, изменения формы губ, недоразвитие челюстей, мелкие и редкие зубы. Радужка глаз имеет густой синий цвет. Фигура тела массивная, кисти рук крупные, с широкими концевыми фалангами. Наблюдаются мышечная гипотония, брадикинезия, атаксия. На высоте лихорадки могут быть эпилептические пароксизмы. Пациенты вялы, но ненадолго могут быть и достаточно активны. Генетическая трансмиссия не установлена, отмечают накопление семейных случаев. Отношение данной патологии к сходным другим остается неясным. Лечение симптоматическое (витамины, ноотропы, логопедические занятия).
25. Синдром Секкеля. Микроцефалия с гипоплазией лица и выступающим носом сочетается с патологиями костной системы, в частности со многими малыми сочленениями. Выраженная умственная задержка и маленький рост. Тип генетической трансмиссии аутосомный рецессивный.
26. Синдром Шегрена-Ларссона (1956). Иногда говорят о синдроме Рада. Сочетание олигофрении с врожденными ихтиозом, эритродермией и спастическими параличами. Наблюдаются эпилептические припадки. Выявляются также перерождение сетчатки, гипертелоризм, дисплазия зубов, возможны гипофизарные нарушения. Умственная отсталость выражена в разной степени, не прогрессирует. Тип генетической трансмиссии аутосомный рецессивный. Специфического лечения не существует.
27. Синдром Смита-Лемли-Опитца. Развернутые вперед ноздри и/или птоз век сопровождаются синдактилией 2-го и 3-го пальцев ноги, а также гипоспадией и крипторхизмом. Выражена умственная ретардация, рост уменьшен. Тип генетической трансмиссии аутосомный рецессивный.
28. Синдром Стерджа-Вебера-Краббе (1879). Наблюдается плоская гемангиома лица, чаще распространяющаяся в зоне иннервации тройничного нерва на одной половине лица с поражением сосудов глазного дна (ангиома, глаукома) на стороне ангиомы. Обнаруживается также гемангиома менингеальной оболочки, она может повлечь эпилептические припадки. Выявляются очаговые обызвествления в мозгу, иногда — асимметрия костей лица и черепа (атрофия костей на одной стороне). Выявляются различные изменения психики, симптомы когнитивного дефицита. Чаще встречается у детей. Тип генетической трансмиссии не установлен.
29. Синдром Тритчер-Коллинза, или мандибулофациальный дизостоз. Наблюдаются гипоплазия скуловых костей в нижней челюсти, скошенные вниз глазные щели, дефект нижнего века и оттопыренные ушные раковины. Может быть умственная ретардация. Тип генетической трансмиссии аутосомный доминантный.
30. Синдром Вильямса (синдром Вильямса-Бойрена, синдром «лицо эльфа», идиопатическая инфантильная гиперкальциемия). Описан в 1952 г. Характерным считается вид лица: полные отвислые щеки, плоское переносье с однотипной для всех больных закругленной формой носа, большой рот с полными губами, особенно нижней, сходящееся косоглазие, эпикант, низко посаженные уши, выступающий затылок. Верхние и нижние веки отечны, глаза голубые с искрящейся радужкой, склеры синеватого цвета.
Мышечная гипотония, опущенные плечи, впалая грудь, круглая спина, Х-образные ноги, плоскостопие. Часто бывают паховая и пупочная грыжи, иногда — врожденный вывих бедра. У старших детей длинные, редкие зубы. Часты врожденные пороки сердца, особенно надклапанный стеноз аорты, стеноз легочной артерии. Длина и масса тела уменьшены. В младенчестве может быть гиперкальциемия. Степень умственной ретардации — от имбецильности до пограничной умственной отсталости. Речь развита неплохо, дети словоохотливы, добродушны, склонны к подражанию и послушанию. Могут быть энурез, страхи, навязчивости, поведение обычно упорядочено. Некоторые пациенты способны к обучению во вспомогательной школе. Тип генетической трансмиссии связан с микроделецией в длинном плече хромосомы 7. Систематического лечения не существует.
31. Болезнь Гиппель-Линдау (1895–1926), ретино-церебеллярный ангиоматоз. Характеризуется множественными ангиобластомами, локализующимися преимущественно в сетчатке и мозжечке, нарушениями развития внутренних органов. Опухоли могут быть доброкачественными (кистомы поджелудочной железы, почек, печени), иногда обнаруживается гипернефрома. Когнитивный дефицит выражен в разной степени. Одновременно могут наблюдаться утомляемость, раздражительность, возбуждение, нарушения поведения, симптомы регрессии. В некоторых случаях возникают симптомы, свойственные опухолям лобной локализации. Тип генетической трансмиссии не установлен.
32. Болезнь Бурневиля (1880), болезнь Прингла-Бурневиля, бугорчатый туберозный склероз головного мозга, эпилойя. Одно из нередких менделирующих заболеваний детей с умственной отсталостью. Часто уже при рождении на коже детей обнаруживаются пятна депигментации овальной, круглой или листовидной формы. Число их может достигать десятков, размеры — до 1 см и более. К 4–5-му году появляются «сальные аденомы», плотные, размером с просяное зерно. Чаще они расположены на лице в виде бабочки или на подбородке. Гистологически такие опухоли включают гиперплазированные сосуды, разрастания фиброзной ткани, незрелые волосяные фолликулы. Реже бывают другие изменения кожи: участки «шагреневой» кожи в пояснично-сакральной области, фибромы вокруг ногтевых лож, очаги гиперпигментации.
На Р-грамме выявляются внутримозговые петрификаты, признаки гидроцефалии, очаги склероза в костях свода черепа, участки разрежения костной ткани, явления гемиатрофии мозга. Встречаются микроцефалия, катаракта, узелковые изменения конъюнктивы, пигментные ретинопатии. В неврологическом статусе выявляется легкая пирамидная недостаточность, реже — параличи и парезы. У детей постарше могут возникать опухоли, особенно часто рабдомиома сердца и опухоли почек.
Умственная ретардация обычно сочетается с эпилептическими припадками. При начале болезни в младенчестве когнитивный дефицит часто соответствует идиотии или глубокой имбецильности. Могут быть состояния психомоторного возбуждения, заторможенности с каталепсией, двигательные стереотипии. Припадки бывают тоническими, фокальными, пропульсивными, иногда возникают серийно.
С началом болезни после трех лет вначале появляются припадки. С их повторением и тем более учащением присоединяются и нарастают симптомы интеллектуального дефицита, психопатоподобного поведения, возникают психотические эпизоды. Тип генетической трансмиссии аутосомный доминантный. Аномальные аллели (их установлено две — в 24-й и в 13-й хромосомах) обладают очень варьирующейся экспрессивностью и очень высокой пенетрантностью. Специфического лечения не существует. Антиконвульсанты малоэффективны, иногда в состоянии эпилептического статуса дети погибают. Считается, что в 80% случаев заболевание связано с мутациями родительских гамет.
33. Синдром Ваарденбурга. Наблюдается латеральное смещение внутреннего угла глаза и впадины. Типичен частичный альбинизм в виде белой пряди волос. Отмечается гетерохромия радужной оболочки, а также витилиго (кожный отрубевидный лишай), может быть глухота. Сведения об умственной отсталости лишены ясности. Тип генетической трансмиссии не установлен.
34. Цереброгепаторенальный синдром Зельвегера. Наблюдаются плоское лицо, высокий лоб, мышечная гипотония. Выявляется гепатомегалия. Рост уменьшен, интеллект не развивается. Дети погибают в раннем младенчестве. Тип генетической трансмиссии аутосомный рецессивный.
35. Синдром трисомии-Х (Джекобс, 1959). У умственно отсталых девочек и женщин встречается с частотой 0,59%. У части лиц с трисомией-X вообще не бывает отклонений в физическом и психическом развитии. У 75% таких людей выявляется неглубокая умственная отсталость. Часто пациенты заболевают шизофренией. У многих пациентов наблюдают задержку физического развития (реже больные имеют высокий рост), а также негрубые даспластические признаки: эпикант, высокое твердое небо, клинодактилия мизинцев. Некоторые пациенты бесплодны из-за недоразвития фолликулов. Диагноз ставится только при цитогенетическом исследовании (обнаруживается дополнительный половой хроматин). Описано немало случаев полисомии-X: тетрасомия (ХХХХ) и пентасомия (ХХХХХ). Степень психического недоразвития выражена при этом грубее и коррелирует с числом дополнительных X-хромосом.
36. Синдром XYY (кариотип 47, XYY). Впервые выявлен в 1960 г. у высокорослых преступников (рост 186 см и выше), однако встречается и у мужчин среднего роста. Наличие добавочной Y-хромосомы иногда ничем себя не обнаруживает. В раннем возрасте пациенты мало пользуются речью, замкнуты, закрыты, не склонны дружить и привязываться, кому-либо симпатизировать. В школьные годы выделяются неустойчивостью внимания, неусидчивостью, неспособностью к длительному умственному напряжению, устойчивым интересам и увлечениям, а также целенаправленной трудовой деятельности.
Отмечаются беспричинные колебания настроения, взрывчатость, импульсивность, агрессивность по малейшему поводу. В то же время пациенты зависимы, внушаемы, склонны подражать делинквентному и асоциальному поведению сверстников. Нравственное развитие часто ограничивается готовностью воспринимать только поощрения и наказания. Совершаются побеги из школы, дома, подростки рано приобщаются к антисоциальным группам, употреблению алкоголя, наркотиков, рано вступают в половые связи. Школьная дезадаптация прерывается отчислением за неуспеваемость, овладеть какими-либо серьезными профессиями многие пациенты оказываются не в состоянии. Неспособны они и к созданию благополучной семьи. У большинства пациентов выявляется дебильность. У части пациентов обнаруживаются диспластические признаки: евнухоидное сложение, неправильное строение зубов, увеличение нижней челюсти, аномальный прикус, девиация коленных и локтевых суставов, радиоульнарный синостоз, расщепление позвоночника. Иногда отмечают повышение уровня андрогенов и лютеинизирующего гормона. Половая функция не нарушена, возможны сексуальные девиации. Диагноз выставляется при цитогенетическом исследовании (в буккальных мазках обнаруживается Y-хроматин), а при изучении кариотипа — дополнительная Y-хромосома. Специфического лечения и эффективной профилактики не существует. Основное значение имеет психокоррекционная работа, рациональная психотерапия.
37. Синдром трисомии-18 Эдвардса. Наблюдаются микростомия, короткие глазные щели, оттопыренные ушные раковины, долихоцефалия. Руки сжаты, второй палец расположен выше третьего. Низкие дужки на кончиках пальцев, короткая грудная клетка. Крипторхизм, врожденные пороки сердца. Рост уменьшен. Выраженная умственная ретардация.
38. Синдром трисомии-13 Патау. Выявляются дефекты глаз, носа, губ, ушей; лоб голопрозенцефального типа. Полидак