Аутосомно-рецессивные заболевания
Аутосомно-рецессивные заболевания проявляются только при гомозиготном носительстве мутантных аллелей. При этом происходит частичная или полная инактивация функции мутантного гена. Одну из мутаций больной ребенок наследует от матери, другую - от отца. Родители больного, будучи сами здоровы, являются гетерозиготными носителми мутации. Вероятность рождения больного ребенка в такой семье в соответствии с законом Менделя составляет 25%. Девочки и мальчики болеют с одинаковой частотой, при этом рождение больного ребенка совершенно не зависит от возраста родителей, очередности беременности и родов. Часто в одной семье может наблюдаться несколько больных сибсов. Две трети здоровых детей в браке гетерозиготных родителей также оказываются гетерозиготными носителями мутации. В браке гетерозиготного носителя рецессивной мутации с супругом, не имеющим мутантного аллеля, все дети будут здоровы, но половина из них окажутся гетерозиготными носителями мутации. Анализ родословных больных с аутосомно-рецессивными заболеваниями показывает, что часто (примерно в 60%) родители таких больных являются родственниками или их предки происходят родом из одного села или района, что так же является косвенным признаком инбридинга.
Муковисцидоз
Самым распространенным аутосомно-рецессивным заболеванием детского возраста среди представителей белой расы является муковисцидоз или кистозный фиброз поджелудочный железы. Впервые заболевание описано в 1938 году американским паталогоанатомом Д. Андерсон. Согласно зарубежной статистике частота муковисцидоза у жителей Западной Европы, в среднем, составляет 1 на 2-3 тысячи новорожденных, в России она меньше - 1 на 3-5 тысяч. Наблюдаются значительные географические и этнические различия по частоте муковисцидоза. В странах Африки и Азии муковисцидоз почти не встречается, тогда как каждый 20-й (5%) житель Западной Европы является гетерозиготным носителем мутации в гене муковисцидоза (CFTR). Такую высокую распространенность мутантных аллелей в гене CFTR связывают как с «эффектом основателя», так и селективным преимуществом гетерозигот, обусловленным устойчивостью к холере, туберкулезу, токсическим формам гриппа. С другой стороны, у гетерозигот вдвое выше частота хронического панкреатита.
Основной патогенетический механизм заболевания - увеличение вязкости секрета, выделяемого слизеобразующими железами бронхов, кишечника, поджелудочной железы, семявыводящих канальцев, сопровождающееся закрытием многих протоков в этих органах. В частности, нарушается естественный процесс очищения бронхов, что приводит к их воспалению. Воспаление сопровождается отеком легких и увеличением продукции аномально вязкого секрета. Повышенная вязкость секрета в желудочно-кишечном тракте сопровождается изменением водно-электролитного компонента панкреатического сока, его сгущением и затруднением выделения в просвет кишечника. В результате нарушается формирование каловых масс с последующей кишечной непроходимостью и происходит фиброзно-кистозное изменение ткани поджелудочной железы.
Минимальными диагностическими симптомами муковисцидоза являются рецидивирующие легочные, чаще всего синегнойные инфекции, нарушение функции кишечника и поджелудочной железы, отставание в физическом развитии. Характерными признаками заболевания считаются большое количество неперевариваемого жира в копрограмме больного и повышение концентрации ионов натрия и хлора при проведении потовой пробы. У некоторых больных уже при рождении наблюдается кишечная непроходимость, обусловленная мекониальным илеусом. Такие больные требуют срочного оперативного вмешательства. Иногда мекониальный илеус у плода с муковисцидозом можно обнаружить при ультразвуковом исследовании уже во 2-3-м триместрах беременности. Выделяют три клинические формы муковисцидоза: легочную (15-20% случаев), кишечную (10%) и смешанную. Описаны также стертые формы заболевания, выявляющиеся у взрослых.
Обследованию на наличие муковисцидоза подлежат следующие группы лиц: (1) больные с рецедивирующими бронхолегочными заболеваниями, синегнойной инфекцией, астмой, аллергозами; (2) лица с заболеваниями желудочно-кишечного тракта (склонные к запорам, колитам, хроническому панкреатиту, рецидивирующей кишечной непроходимости; новорожденные с мекониальным илеусом, перитонитом; дети с большим животом и низкой массой тела (при нормальном аппетите);больные циррозом печени неясного генеза; (3) мужчины с бесплодием после исключения других причин (у 98% мужчин больных муковисцидозом имеется сужение или атрезия семявыводящего протока). Муковисцидоз входит в программу неонатального скрининга новорожденных.
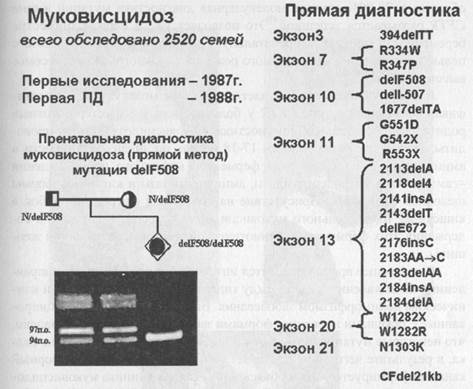
Ген муковисцидоза был картирован в 1985 году в длинном плече хромосомы 7 в области 7q31.2. В 1989 году он был идентифицирован, и это открытие сопровождалось одновременной публикацией в одном из самых престижных журналов мира («Science» - Наука) трех статей. Первая статья была посвящена самому гену муковисцидоза. Группе канадских исследователей под руководством Л. Ч. Тсуи удалось выделить и определить нуклеотидную последовательность кДНК гена муковисцидоза (CFTR). Были определены экзон-интронные границы и регуляторные области гена. Вторая статья была посвящена белку, кодируемому геном CFTR и являющимся первичным биохимическим дефектом у больных муковисцидозом. Оказалось, что этот трансмембранной белок, локализованный на апикальных мембранах экзокринных желез эпителия, участвует в регуляции проводимости ионов хлора и сам выполняет функции хлорного канала. В третьей статье был проведен анализ мутаций в гене CFTR. Как только становится известна нуклеотидная последовательность кодирующей области гена, сразу же можно задаться вопросом, а что случилось у больного, чем ген больного человека отличается от нормального гена? Используя нормальную кДНК гена CFTR в качестве ДНК-зонда, удалось выделить мутантную кДНК из эпителия бронхов девочки, больной муковисцидозом. На следующем этапе была определена полная нуклеотидная последовательность этой кДНК. Оказалось, что больная девочка гомозиготна по специфической мутации в гене CFTR - делеции трех нуклеотидов в 10-м экзоне, сопровождающейся отсутствием фенилаланина в 508 положении соответствующего белка - delF508. Частота этой мутации у больных муковисцидозом в Канаде, Северной Америке и Северной Европе достигает 80%. Мутация delF508 легко диагностируется методом ПЦР и электрофоретического разделения ампли-фицированных фрагментов ДНК. На рис.57 представлены мутации в гене CFTR, диагностируемые в лаборатории пренатальной диагностики наследственных и врожденных заболеваний ИАГ им. Д. О. Отта РАМН, возглавляемой чл-корр. РАМН проф. В. С. Барановым, указано количество обследованных семей больных муковисцидозом и дан пример пренатальной диагностики delF508. Рисунок 57. Диагностика мутаций в гене CFTR у больных муковис-цидозом (данные 2002 г.).
В настоящее время у больных муковисцидозом идентифицировано более 1000 разных мутаций в гене CFTR, главным образом, миссенс-типа. Однако самой распространенной остается delF508. Ее частота у больных муковисцидозом в разных популяциях варьирует от 30% до 80%. В Европе наблюдается определенный градиент распространения этой мутации с севера на юг: в Дании ее частота достигает 85%, в Италии снижается до 50% и в Турции - до 20-30%. В славянских популяциях частота delF508 среди больных муковисцидозом составляет около 50%. К мажорным мутациям относятся миссенс-мутации W1272X (встречается более чем в 30% случаев у больных муковисцидозом, принадлежащих этнической группе евреев ашкенази), G542X, G551D, R117H, R334W и др. Таким образом, в 70-80% случаев молекулярная диагностика мутаций в гене CFTR оказывается успешной. Это позволяет уже в первом триместре беременности проводить пренатальную диагностику муковисцидоза с целью предупреждения повторного рождения больного ребенка в семье высокого риска.
В тех случаях, когда не удается провести молекулярную идентификацию мутаций в гене CFTR у больного или у его гетерозиготных родителей, пренатальная диагностика муковисцидоза может проводиться при сроке беременности 17-18 недель по анализу активности в амниотической жидкости ряда ферментов кишечного происхождения -гамма-глютамилтранспептидазы, аминопептидазы и кишечной формы щелочной фосфатазы. Присутствие на этом сроке слизистых пробок в кишечнике плода больного муковисцидозом приводит к снижению содержания этих ферментов в амниотической жидкости беременной женщины.
В настоящее время проводятся интенсивные исследования, направленные на выявление связей между типами мутаций в гене CFTR и клиническим полиморфизмом заболевания. Выявлены мутации, ассоциированные с тяжелыми и мягкими формами заболевания. Было установлено, что некоторые мутации, в том числе delF508, нарушают процессинг белка, в результате чего он не достигает апикальной мембраны и хлорный канал не формируется. Этим объясняется тяжелая клиника муковисцидоза при подобных нарушениях. Другие мутации (R117H, R334W, R347P), выявленные при более мягких формах муковисцидоза, не затрагивают процессинг белка, хлорный канал формируется, но работает менее интенсивно. Так, например миссенс-мутация Rl 17Н обнаружена у мужчин, страдающих бесплодием в силу закупорки семявыводящих канальцев. При этом клиника муковисцидоза у таких пациентов, как правило, отсутствует или очень стерта. То есть, у носителей мутации R117H вязкость аномального секрета, выделяемого экзокринными железами эпителия, повышена настолько незначительно, что это не приводит к аномальным процессам в легких, поджелудочной железе или в кишечнике, но это повышение достаточно для формирования непроходимости семявыводящих канальцев (vas deferens).
С использованием техники трансгеноза в различных лабораториях США и Великобритании были сконструированы модельные линии мышей с мутациями в гене муковисцидоза, в том числе и такими, которые были идентифицированы у больных. Показано, что различные мутации по-разному влияют на фенотип животных. У мышей некоторых трансгенных линий отмечено преимущественное поражение легких, тогда как в других линиях - поджелудочной железы и кишечника. В одной линии наблюдали гибель большого числа зародышей от причин, сходных с ме-кониальным илеусом. Таким образом, эти линии представляют собой идеальные модели не только для изучения молекулярных основ патогенеза муковисцидоза, но и испытания различных программ терапии этого тяжелого заболевания.
В настоящее время разработаны эффективные методы этиопато-генетического лечения, позволяющие значительно увеличить продолжительность жизни больных муковисцидозом. Лечение больных муко-висцидозом показано проводить в специализированных региональных центрах. Общая схема лечения включает применение муколитических средств, антимикробных препаратов, ферментов поджелудочной железы, витаминов, в сочетании с применением кинезо- и физиотерапии. Ведение больных муковисцидозом проводится согласно национальным рекомендациям.
Наследственные болезни обмена
По аутосомно-рецессивному типу наследуются болезни обмена - одна из наиболее многочисленных и хорошо изученных групп моногенных заболеваний человека. Наследственные болезни обмена обусловлены нарушением каталитической функции различных ферментов. Подобные нарушения часто сопровождаются накоплением веществ, предшествующих ферментативному блоку, и дефицитом конечных продуктов реакции. Частоты наследственных болезней обмена колеблются в очень широких пределах от 1:2-3 тысячи новорожденных до 1:105-106, причем для многих подобных заболеваний характерны выраженные различия по частотам встречаемости в разных этнических группах и популяциях. Как правило, наследственные болезни обмена это тяжелые состояния, клинические проявления которых очень разнообразны. Часто они включают задержку психомоторного развития, судорожный синдром, миопатию, скелетные аномалии, рецидивирующие каматозные состояния, кетоацидоз, гепатоспленомегалию, мальабсорбцию, атаксию, синдром внезапной смерти.
Выделяют следующие группы наследственных болезней обмена: нарушения обмена аминокислот - аминоацидопатии (альбинизм, фенил-кетонурия, тирозинемия, алкаптонурия, гомоцистинурия, гистидинемия, гипервалинемия, гиперлизинемия, тирозиноз и др.); углеводов - глю-козурии (галактоземия, гликогенозы, фруктозурия и др.); липидов (ги-перхолестеринемия, гиперлипидемия, ганглиозидозы, сфинголипидозы, лейкодистрофии и др.); гликозаминогликанов (мукополисахаридозы); стероидных и глюкокортикоидных гормонов (адреногенитальный син  дром); пуринов и пиримидинов (ксантинурия и др.); билирубина (синдром Криглера-Найяра); металлов (гемохроматоз, Вильсона-Коновалова); порфирина (эритропоэтическая и другие порфирии.
дром); пуринов и пиримидинов (ксантинурия и др.); билирубина (синдром Криглера-Найяра); металлов (гемохроматоз, Вильсона-Коновалова); порфирина (эритропоэтическая и другие порфирии.
Представляем более подробное описание наиболее известных наследственных болезней обмена, с которыми врач может встретиться в своей повседневной практике. Знакомство с наследственными болезнями обмена необходимо еще и потому, что для некоторых из этих заболеваний проводятся биохимические скрининги среди новорожденных с целью их ранней диагностики и своевременного лечения.
2.6.3.1. Фенилкетонурия
Общими нарушениями при наследственных дефектах обмена ами нокислот являются аминоацидурия (выделение аминокислот с мочей) и ацидоз тканей. Наиболее распространенные аминоацидопатии обусловлены дефектами метаболизма двух аминокислот - фенилаланина и тирозина. На рис.61 изображены биохимические превращения фенилаланина и тирозина и основные метаболические блоки на их пути.

Рисунок 61. Обмен фенилаланина и тирозина
Гиперфенилаланинемии - это группа генетически гетерогенных аутосомно-рецессивных заболеваний, обусловленных нарушением метаболизма фенилаланина. В основе патогенеза гиперфенилаланинемии лежит накопление в крови фенилаланина и продуктов его утилизации: фенилпировиноградной, фенилмолочной и фенилуксусной кислот, оказывающих токсический эффект на различные органы и ткани, в первую очередь на головной мозг.
Фенилаланин - незаменимая аминокислота, которая не синтезируется в организме, а поступает с пищей. С помощью фермента фенилаланин-гидроксилазы, экспрессирующегося в печени, фенилаланин превращается в тирозин. Фенилкетонурия, наиболее частая и злокачественная форма гиперфенилаланинемии. Заболевание, впервые описанное Феллингом в 1934 году, обусловлено наследственной недостаточностью фенилала-нингидроксилазы. В нашей стране исследования по фенилкетонурии детально впервые были проведены московским генетиком и психиатром М. Г. Блюминой в 1970-е годы. Частота фенилкетонурии составляет 1 на 8-10 тысяч новорожденных, частота гетерозиготного носительства - 1: 50-100 человек. В настоящее время описано еще семь более редких наследственных гиперфенилаланинемии, обусловленных мутациями в генах, кодирующих другие ферменты метаболизма фенилаланина.
В основе патогенеза фенилкетонурии лежит накопление фенилаланина и его побочных продуктов, оказывающих токсичное действие на мозг и другие органы больного. Ферментативный блок превращения фенилаланина сопровождается уменьшением синтеза медиаторов центральной нервной системы - дофамина и диоксифенилаланина, а также дефицитом конечного продукта реакции - меланина. Ведущим симптомом болезни является отставание умственного развития, достигающее у большей части больных степени имбецильности или идиотии. Уже с первых недель жизни ребенка наблюдаются повышенная возбудимость, повышение рефлексов, мышечная ригидность и судорожный синдром. Одним из первых неспецифических проявлений заболевания может быть повторяющаяся рвота. В 80-90% наблюдений у детей выражен дефект пигментации, обусловленный дефицитом меланина. Большинство из них блондины с голубыми глазами и светлой кожей. Нередки мокнущие экземы и дерматиты. Всем новорожденным проводится обязательное централизованное специальное скринирующее исследование для выявления среди них больных фенилкетонурией. Иллюстрацией результативности Налаженный скрининг совсем не исключает проведение диагностических мероприятий по выявлению фенилкетонурии среди групп детей повышенного риска. Таковыми являются умственно неполноценные дети, находящиеся в специализированных учреждениях; дети, отстающие в умственном развитии, с поражениями кожных покровов; а также братья и сестры больных фенилкетонурией.
Больные фенилкетонурией являются гомозиготными носителями мутаций в гене РАН, ответственном за синтез фенилаланингидроксила-зы. При нарушении активности указанного фермента концентрация фе-нилаланина в крови и во многих органах больного резко повышена. В частности, накопление этой аминокислоты в головном мозге вызывает интоксикацию и гибель нервных клеток с соответствующими для этого заболевания клиническими проявлениями.
Ген РАН идентифицирован, и он находится на 12 хромосоме (12q22-24). Определены спектры наиболее частых мутаций в гене РАН, которые оказались разными для разных этнических групп. В странах Восточной Европы (Польше, Белоруссии, России), где фенилкетонурия встречается с высокой частотой, мажорными являются мутации R408W (ее частота у больных достигает 60%), R158Q и др. Наличие мажорных мутаций значительно облегчает молекулярную диагностику заболевания и выявление гетерозиготных носителей мутантных аллелей. Это крайне важно для медико-генетического консультирования членов семьи больного и прогнозирования фенилкетонурии у их потомства. На рис.63 представлен пример пренатальной диагностики мутации R408W в гене РАН методом рестрикционного анализа, указаны количество обследованных семей и мутации в гене РАН, диагностируемые в лаборатории пренатальной диагностики наследственных и врожденных заболеваний НАГ им. Д. О. Отта РАМН (зав. лаб. чл-корр. РАМН проф. В. С. Баранов, г. Санкт-Петербург). Подобная диагностика может быть проведена по высушенному на фильтровальной бумаге пятну крови больного. Лечение больных заключается в исключении из питания фенилала-нина путем применения специфической безфенилаланиновой диеты. Это малобелковые продукты под названием амилофены и лечебные продукты: афинелак, тетрафен (Россия). Не можем не отметить, что одними из первых в нашей стране диетическое лечение больных фенилкетонурией предложили и разработали в 60-е годы прошлого столетия отечественные ученые Института экспериментальной медицины РАМН в Санкт-Петербурге член-корр. АМН СССР, профессор С. А. Нейфах (1909-1998) и доктор медицинских наук А. М. Шапошников. Предпочтительно на спецализированной диете больному фенилкетонурией находится в течение всей жизни. После второй декады и стабилизации состояния и содержания фенилаланина в крови возможно расширение диеты.
Особую проблему представляют беременные женщины, ранее находившиеся на безфенилаланиновой диете, так называемая «материнская фенилкетонурия». Их плоду угрожает фенилаланиновая эмбриопатия, которая проявляется микроцефалией, пороками сердца, пренатальной гипоплазией и умственной отсталостью. У таких женщин беременность должна планироваться, и с первых дней ей необходимо соблюдать без-фенилаланиновую диету.
Галактоземия
Наиболее известной наследственной болезнью углеводного обмена являются галактоземия. Заболевание впервые описано А. Ресом в 1908 году. Его частота в разных популяциях составляет 1 на 15000-20000 новорожденных. Болезнь обусловлена недостаточностью или отсутствием ферментов, участвующих в метаболизме галактозы (рис.64). Ведущим из них является галактозо-1 -фосфатуридилтрансфера (Г 1 ФУТ). При недостаточности этого фермента, обусловленной присутствием мутаций в гене GALT, развивается классическая форма заболевания первого типа. Недостаточность галактокиназы, обусловленная мутациями в гене GALK1, расположенном в области 17q24, является причиной развития второго типа заболевания. В основе развития третьего типа заболевания лежит недостаточность фермента галактозоепимеразы, связанная с мутациями в гене GALE, локализованном в области 1р36-р35.
При всех формах заболевания происходит метаболический блок превращений галактозы и галактозо-1 -фосфата. При избыточной концентрации эти метаболиты оказывают токсическое действие на ткани мозга, печени, почек и кишечника. Ингибируется бактерицидная активность лейкоцитов, что приводит к развитию септических осложнений. При очень высоком содержании галактозы она начинает метаболизироваться по побочному пути с образованием сахарного спирта - галактиола. Увеличение содержания галактиола может приводить к разрыву зонулярных волокон хрусталика и возникновению катаракт.
Остановимся более подробно на классической форме галактоземии. Клинически болезнь проявляется в следующем: через несколько дней после первого приема молочной пищи появляются рвота, диарея, желтуха (увеличение уровня прямого билирубина), гепатомегалия, диспепсия, асцит, гипотрофия, гемолитические проявления. У большинства больных в первые месяцы жизни формируется катаракта, а также наблюдается отставание психического развития. У больных в моче выявляются галактозурия, протеинурия и аминоацидурия. Все эти симптомы прогрессируют на фоне получения ребенком женского и/или коровьего молока, в которых содержится галактоза Биохимическая диагностика заболевания базируется на измерении активности Г1ФУТ в эритроцитах. ДНК диагностика основана на определении мутаций в гене GALT, локализованном в длинном плече хромосомы 9 в области 9ql3. В европейской популяции наиболее частой является мутация Q188R, диагностируемая у 70% больных. Диагностику галактеземии можно проводить на доклинической стадии при биохимическом скрининге новорожденных.
Рисунок 64. Метаболические блоки превращения галактозы, приводящие к галактоземии.

|
При установлении диагноза галактоземии ребенка необходимо переводить на безлактозную диету - соевое молоко в сочетании с безгалак-тозными белковыми гидролизатами, в частности, гидролизатом казеина. Однако, в отличие от фенилкетонурии, это не всегда приводит к полному исчезновению симптомов.
Адреногенитальный синдром
Примером наследственного нарушения обмена стероидных и глю-кокортикоидных гормонов является адреногенитальный синдром или врожденная гиперплазия коры надпочечников. Заболевание впервые описано в 1886 году J. Phillips. Частота адреногенитального синдрома по данным разных авторов составляет 1 на 5000 - 15000 новорожденных, частота гетерозиготного носительства 1 на 20-50 человек. Только в середине прошлого века были раскрыты патогенез и характер гормональных нарушений при этом заболевании. При адреногенитальном синдроме наблюдается гиперплазия коры надпочечников, что ведет к патологической концентрации кортизола в ответ на стимуляцию адренокортикотропно-го гормона. Это связано с уменьшением или отсутствием ферментативной активности на некоторых стадиях синтеза стероидов. За дефицитом каждого из ферментов следует специфическая для данного нарушения клиническая картина заболевания. В настоящее время описано 5 клинических вариантов адреногенитального синдрома.
Около 90% всех случаев заболевания относится к дефициту ключевого фермента биосинтеза гормонов коры надпочечников - 21- гидроксилазы, обусловленному мутациями в гене CYP21A2 (так называемый синдром дефицита 21-гидроксилазы - СД21-Г). В редких случаях у больных идентифицированы мутации в генах, кодирующих другие ферменты биосинтеза глюкокортикоидов и минералокортикоидов. При дефиците 21 -гидроксилазы наблюдается уменьшение уровня кортизола в плазме крови, вызванное нарушением превращения 17-гидрооксипролина в 11-дезоксикортизол. Это в свою очередь вызывает избыточную секрецию адренокортикотропного гормона и далее повышение продукции предшественников кортизола, андрогенов и половых стероидов. Подобные дефекты гормонального метаболизма приводят, прежде всего, к нарушениям развития в области половой сферы. Заболевание характеризуется значительной клинической гетерогенностью. Классические варианты делятся на тяжелую сольтеряющую и более легкую простую вирильную формы. В первом случае у больных наблюдаются выраженные нарушения солевого обмена в виде гипонатриемии и ги-перкалиемии, которые у новорожденных или в раннем неонатальном периоде могут приводить к летальным сольтеряющим кризам. Нарушения солевого метаболизма сопровождаются различными пороками развития наружных половых органов, выраженность которых зависит от степени остаточной активности дефектного фермента. При простой форме заболевания, которая клинически проявляется в виде изолированной вирилизации наружных половых органов, не наблюдается грубых нарушений гормонального метаболизма, так как происходит компенсация частичной недостаточности 21-гидроксилазы. Неклассические или взрослые варианты включают различные нарушения полового созревания в подростковом или пубертатном возрасте в виде гирсутизма, акне, аменореи, бесплодия. В некоторых случаях подобные патологические состояния у больных женщин развиваются после рождения ребенка. Выделяют также позднюю неклассическую и латентную бессимптомную формы заболевания.

|
Рисунок 67. Метаболизм стероидов и генетические варианты адреногенитального синдрома.
-
Простая форма составляет 1/3 всех случаев заболевания. У девочек с рождения наблюдаются признаки маскулинизации вплоть до трудности определения пола ребенка, что диктует немедленное исследование кариотипа или хотя бы полового хроматина. У мальчиков вирильная форма диагностируется с 5 лет и старше при появлении признаков преждевременного полового развития. Опережение физического и полового развития может быть причиной неадекватного поведения, психических нарушений, преимущественно эмоционально-волевой сферы. У взрослых больных наблюдается олигоспермия (пониженный объем эякулята) и бесплодие. У девочек - гиперпигментация в области гениталий и грудных желез.
При сольтеряющей форме кроме вышеуказанных симптомов уже в периоде новорожденности ребенка наблюдаются срыгивания, упорная рвота, потеря веса, признаки эксикоза; характерно развитие коллапто-идных кризов с цианозом и бледностью, потливостью, потеря сознания, иногда судороги. Характерна внезапность возникновения кризов, длительность которых может варьировать от нескольких минут до получаса. Необходимо отметить, что данные кризы, протекающие с недостаточностью кровообращения, могут приводить к гибели больного. В крови отмечается гиперкалиемия, метаболический ацидоз, может быть гипогликемия.
При третьей поздней неклассической форме у новорожденных девочек отсутствуют признаки вирилизации. Первые проявления патологии манифестируют в подростковом возрасте. Для девочек характерно ранние менархе (возраст наступления первой менструации) до развития молочных желез, гирсутизм, маскулинное телосложение. Для мальчиков - ускорение костного возраста с ранним закрытием ростовых зон, преждевременное оволосение лобковой области. Клинические проявления четвертой латентной формы заболевания отсутствуют, хотя, как и при других формах, в сыворотке крови наблюдается умеренное повышение предшественников кортизола, включая 17-гидрооксипролин.
Ген CYP21A2 расположен в коротком плече хромосомы 6 (6р21.3) в области локализации HLA-генов класса III. Эта область отличается высокими рекомбиногенной и мутагенной активностями. В частности, в непосредственной близости от гена CYP21A2 расположен псевдо-ген CYP21A1P, неактивный вследствие наличия целой серии мутаций. Спектр мутаций в гене CYP21A2 у больных адреногенитальным синдромом хорошо изучен. Широкое распространение в разных популяциях имеют крупные делеции, включающие весь ген CYP21A2, мутации, гомологичные тем, которые присутствуют в псевдогене CYP21A1P и «химерные» конструкции, состоящие из фрагментов гена CYP21A2 и псевдогена CYP21A1P. На рис.68 представлены мутации в гене CYP21A2, диагностируемые в лаборатории пренатальной диагностики наследственных и врожденных заболеваний ИАГ им. Д. О. Отта РАМН (зав. лаб. чл-корр АМН проф. В. С. Баранов), указано количество обследованных семей юльных и представлены примеры диагностики delA2 в гене CYP21A2. При различных классических вариантах адреногенитального синдрома - сольтеряющей и простой - выявляются разные мажорные мута-(ии. В первом случае самой частой является делеция всего гена (delA2), [рисутствующая почти у половины отечественных больных, в то время:ак во втором случае на первое место выступает сплайсинговая мутация 28%). Среди пациентов с подозрением на неклассический вариант врожденной гиперплазии коры надпочечников мутации идентифицируются [римерно в 10% хромосом больных. Интересно отметить, что у больных дреногенитальным синдромом идентифицированы два типа химерных онструкций между геном CYP21A2 и псевдогеном CYP21A1P, причем дна из них является полиморфной, так как ее частота в отечественной популяции достигает 6%. Но еще выше - около 15% - эта частота оказывается среди больных с неклассической формой врожденной гиперплазии коры надпочечников, подтвержденной биохимическим тестированием. При этом общая частота двух «химерных» конструкций в хромосомах этой группы больных составляет 25%. Еще более высокая частота полиморфной «химерной» конструкции обнаруживается в хромосомах женщин с идиопатической андрогениза-цией - 28,5%, а суммарная частота обеих «химерных» конструкций в хромосомах этих больных составляет 38%, что, безусловно, указывает на ассоциацию этого состояния с присутствием гетерозиготных «химерных» конструкций между геном CYP21A2 и псевдогеном CYP21A1P.
В медико-генетических центрах страны проводится скрининг среди новорожденных для раннего выявления больных адреногенитальным синдромом. При всех формах адреногенитального синдрома применяется заместительная терапия минерало-, глюкокортикоидными препаратами, в зависимости от формы заболевания - симптоматическая терапия.