В лабораторной работе проводился расчёт молекулярных характеристик гомоядерной двухатомной молекулы С2 полуэмпирическим методом MNDO.
С помощью программы HyperChem была собрана молекула С2 и скорректирована её геометрия. Полуэмпирическим методом MNDO был произведён расчёт квантово-химический расчёт с оптимизацией геометрии.
Точечная группа симметрии молекул - C∞h
Полная энергия = -72.3249 ккал/моль
Разность энергий ВЗМО и НСМО:???
Межъядерное расстояние: 1.3828Å
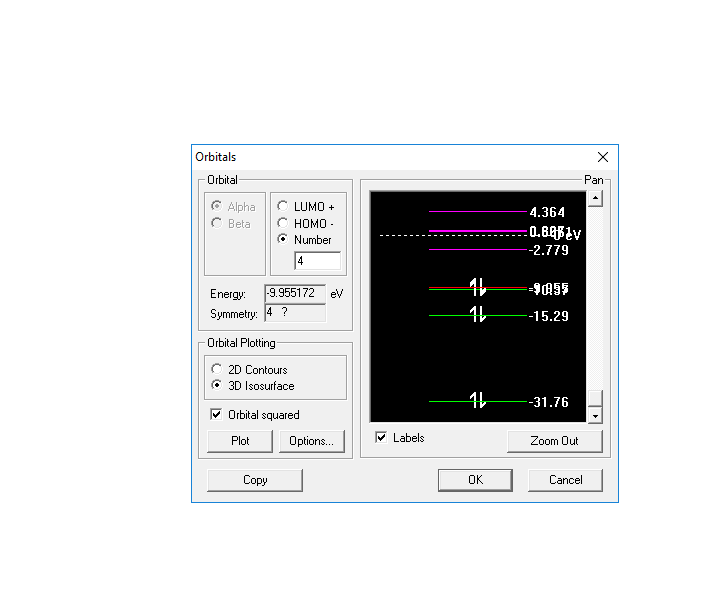
Энергия ВЗМО = эВ Вид симметрии -1σg
Энергия НСМО = 4.364эВ Вид симметрии - 2σu
Энергетическая диаграмма С2
Молекула С2



 Верхняя незаселенная орбиталь
Верхняя незаселенная орбиталь
Нижняя незаселенная орбиталь


HyperChem log start -- Mon Oct 29 17:14:10 2018.
Geometry optimization, SemiEmpirical, molecule = C2.hin.
MNDO
PolakRibiere optimizer
Convergence limit = 0.0010000 Iteration limit = 50
Accelerate convergence = YES
Optimization algorithm = Polak-Ribiere
Criterion of RMS gradient = 0.0010 kcal/(A mol) Maximum cycles = 30
RHF Calculation:
Singlet state calculation
Number of electrons = 8
Number of Double Occupied Levels = 4
Charge on the System = 0
Total Orbitals = 8
Starting MNDO calculation with 8 orbitals
E=-72.3249 kcal/mol Grad=0.000 Conv=YES(3 cycles 11 points) [Iter=1 Diff=0.00000]
Eigenvalues (eV) and Eigenvectors
Mol. Orbital 1 2 3 4 5 6
Symmetry: 1 SIG 1 SIU 3? 4? 5? 6?
Eigenvalue -31.75572 -15.29378 -10.36702 -9.95517 -2.77937 0.56511
S C 1 -0.65371 0.63309 -0.00000 -0.26957 0.00000 0.00000
Px C 1 0.00000 0.00000 -0.52024 -0.00000 -0.47890 0.52024
Py C 1 -0.26957 -0.31495 -0.00000 0.65371 -0.00000 0.00000
Pz C 1 0.00000 0.00000 0.47890 -0.00000 -0.52024 -0.47890
S C 2 -0.65371 -0.63309 -0.00000 -0.26957 -0.00000 -0.00000
Px C 2 -0.00000 0.00000 -0.52024 0.00000 -0.47890 -0.52024
Py C 2 0.26957 -0.31495 -0.00000 -0.65371 -0.00000 -0.00000
Pz C 2 -0.00000 0.00000 0.47890 0.00000 -0.52024 0.47890
Mol. Orbital 7 8
Symmetry: 7? 8?
Eigenvalue 0.82697 4.36389
S C 1 -0.00000 -0.31495
Px C 1 0.47890 0.00000
Py C 1 0.00000 -0.63309
Pz C 1 0.52024 0.00000
S C 2 -0.00000 0.31495
Px C 2 -0.47890 0.00000
Py C 2 -0.00000 -0.63309
Pz C 2 -0.52024 0.00000
ENERGIES AND GRADIENT
Total Energy = -5630.0720024 (kcal/mol)
Total Energy = -8.972089729 (a.u.)
Binding Energy = -72.3248804 (kcal/mol)
Isolated Atomic Energy = -5557.7471220 (kcal/mol)
Electronic Energy = -8728.6079422 (kcal/mol)
Core-Core Interaction = 3098.5359398 (kcal/mol)
Heat of Formation = 269.4551196 (kcal/mol)
Gradient = 0.0014640 (kcal/mol/Ang)
MOLECULAR POINT GROUP
D*H
EIGENVALUES(eV)
Symmetry: 1 SIG 1 SIU 3? 4? 5?
Eigenvalue: -31.755717 -15.293782 -10.367018 -9.955172 -2.779367
Symmetry: 6? 7? 8?
Eigenvalue: 0.565114 0.826967 4.363886
ATOMIC ORBITAL ELECTRON POPULATIONS
AO: 1 S C 1 Px C 1 Py C 1 Pz C 2 S C
1.801611 0.541302 1.198389 0.458698 1.801611
AO: 2 Px C 2 Py C 2 Pz C
0.541302 1.198389 0.458698
NET CHARGES AND COORDINATES
Atom Z Charge Coordinates(Angstrom) Mass
x y z
1 6 0.000000 0.00000 -0.69140 0.00000 12.01100
2 6 -0.000000 -0.00000 0.69140 -0.00000 12.01100
ATOMIC GRADIENTS
Atom Z Gradients(kcal/mol/Angstrom)
x y z
1 6 -0.00000 0.00254 -0.00000
2 6 0.00000 -0.00254 0.00000
Dipole (Debyes) x y z Total
Point-Chg. 0.000 -0.000 0.000 0.000
sp Hybrid -0.000 -0.000 0.000 0.000
pd Hybrid 0.000 0.000 0.000 0.000
Sum -0.000 -0.000 0.000 0.000
HyperChem log stop -- Mon Oct 29 17:14:43 2018.
Часть 2. Изучение гетероядерной двухатомной молекулы NO.
В лабораторной работе проводился расчёт молекулярных характеристик гетероядернойй двухатомной молекулы NO полуэмпирическим методом MNDO.
С помощью программы HyperChem была собрана молекула NO и скорректирована её геометрия. Полуэмпирическим методом MNDO был произведён расчёт квантово-химический расчёт с оптимизацией геометрии.
Точечная группа симметрии молекул –C∞v
Полная энергия =-172.7174 ккал/моль
Разность энергий ВЗМО и НСМО:
11.791 эВ= 1.889*10-18 Дж/моль= 1.2655*10-8 а.е.м
Межъядерное расстояние: 1.12447Å
Заряды на атомах = 0.025251 и -0.25251
Дипольный момент = 0.174 Д
Энергия ВЗМО = -4.545 эВ Вид симметрии -3σ
Энергия НСМО = 7.246эВ Вид симмерии-4σ
Энергетическая диаграмма NO


 Верхняя незаселенная орбиталь
Верхняя незаселенная орбиталь

 Нижняя незаселенная орбиталь
Нижняя незаселенная орбиталь
 Заселенная орбиталь
Заселенная орбиталь

HyperChem log start -- Mon Oct 29 17:33:53 2018.
Geometry optimization, SemiEmpirical, molecule = NO.hin.
MNDO
PolakRibiere optimizer
Convergence limit = 0.0010000 Iteration limit = 50
Accelerate convergence = YES
Optimization algorithm = Polak-Ribiere
Criterion of RMS gradient = 0.0010 kcal/(A mol) Maximum cycles = 30
RHF Calculation:
Doublet state calculation
The half-electron approximation will be used
Number of electrons = 11
Number of Double Occupied Levels = 5
Number of Single Occupied Levels = 1
WARNING message received from node=0:
The optimized geometry may not correspond to the geometry with the minimum energy because of the half electron approximation.
Charge on the System = 0
Total Orbitals = 8
Starting MNDO calculation with 8 orbitals
E=-172.7174 kcal/mol Grad=0.000 Conv=YES(3 cycles 15 points) [Iter=1 Diff=0.00000]
Eigenvalues (eV) and Eigenvectors
Mol. Orbital 1 2 3 4 5 6
Symmetry: 1 SI 2 SI 1 PI 1 PI 3 SI 2 PI
Eigenvalue -46.67905 -24.92629 -17.23081 -17.00085 -16.76683 -4.54511
S N 1 -0.48208 0.68629 -0.00000 0.00000 -0.48629 -0.00000
Px N 1 -0.00000 -0.00000 0.57896 -0.13549 0.00000 0.78349
Py N 1 -0.33665 -0.04072 -0.00000 0.00000 0.62901 -0.00000
Pz N 1 0.00000 0.00000 0.13415 0.58472 -0.00000 0.18155
S O 2 -0.75974 -0.57804 0.00000 0.00000 -0.18160 0.00000
Px O 2 0.00000 -0.00000 0.78349 -0.18055 -0.00000 -0.57896
Py O 2 0.27760 -0.43957 -0.00000 0.00000 -0.57869 0.00000
Pz O 2 -0.00000 0.00000 0.18155 0.77919 0.00000 -0.13415
Mol. Orbital 7 8
Symmetry: 2 PI 4 SI
Eigenvalue 0.13067 7.24584
S N 1 0.00000 -0.24521
Px N 1 0.18055 -0.00000
Py N 1 -0.00000 -0.69953
Pz N 1 -0.77919 0.00000
S O 2 0.00000 0.23598
Px O 2 -0.13549 -0.00000
Py O 2 0.00000 -0.62836
Pz O 2 0.58472 0.00000
ENERGIES AND GRADIENT
Total Energy = -12174.4689438 (kcal/mol)
Total Energy = -19.401248815 (a.u.)
Binding Energy = -172.7174088 (kcal/mol)
Isolated Atomic Energy = -12001.7515350 (kcal/mol)
Electronic Energy = -19259.7204708 (kcal/mol)
Core-Core Interaction = 7085.2515270 (kcal/mol)
HE Energy Correction = -65.4415466 (kcal/mol)
Heat of Formation = -0.1584088 (kcal/mol)
Gradient = 0.0000098 (kcal/mol/Ang)
MOLECULAR POINT GROUP
C*V
EIGENVALUES(eV)
Symmetry: 1 SI 2 SI 1 PI 1 PI 3 SI
Eigenvalue: -46.679051 -24.926288 -17.230814 -17.000847 -16.766833
Symmetry: 2 PI 2 PI 4 SI
Eigenvalue: -4.545109 0.130670 7.245839
ATOMIC ORBITAL ELECTRON POPULATIONS
AO: 1 S N 1 Px N 1 Py N 1 Pz N 2 S O
1.879740 1.320951 1.021304 0.752754 1.888628
AO: 2 Px O 2 Py O 2 Pz O
1.628093 1.210328 1.298202
NET CHARGES AND COORDINATES
Atom Z Charge Coordinates(Angstrom) Mass
x y z
1 7 0.025251 -0.00000 -0.63697 0.00000 14.00700
2 8 -0.025251 0.00000 0.48750 -0.00000 15.99900
ATOMIC GRADIENTS
Atom Z Gradients(kcal/mol/Angstrom)
x y z
1 7 0.00000 0.00002 -0.00000
2 8 -0.00000 -0.00002 0.00000
Dipole (Debyes) x y z Total
Point-Chg. -0.000 -0.136 0.000 0.136
sp Hybrid 0.000 0.310 -0.000 0.310
pd Hybrid 0.000 0.000 0.000 0.000
Sum 0.000 0.174 -0.000 0.174
HyperChem log stop -- Mon Oct 29 17:34:40 2018.