ХРОМОСОМНЫЕ БОЛЕЗНИ.
Хромосомные болезни — это большая группа врожденных наследственных заболеваний, которые клинически характеризуются наличием множественных пороков развития, а в качестве этиологической основы имеют численные или структурные аномалии хромосом.
В основе хромосомных заболеваний, обусловленных изменением структуры отдельных хромосом, лежат следующие механизмы: транслокация (обмен сегментами различных хромосом); делеция (утрата части хромосомы); дупликация (удвоение сегмента хромосомы); инверсия (разрыв хромосомы в двух местах и поворот этого участка на 180°).
Хромосомные аномалии имеют широкий спектр клинических проявлений. Они могут быть причиной врожденных пороков развития, повторных самопроизвольных абортов, случаев мертворождения, неонатальной смертности и бесплодия.
1.Аутосомные трисомии.
2.Аномалии числа половых хромосом.
3.Структрурные аномалии хромосом.
АУТОСОМНЫЕ ТРИСОМИИ
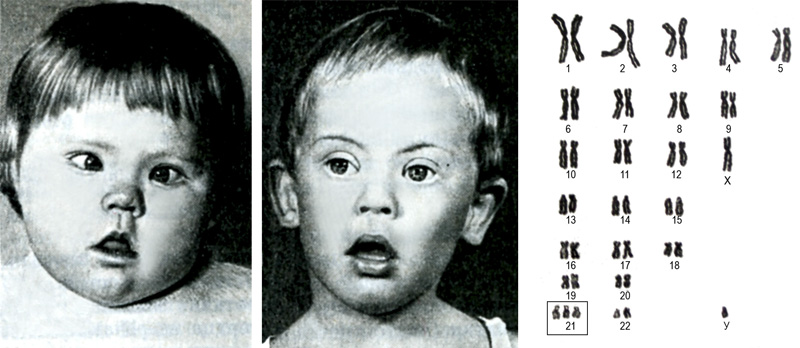
1. Синдром Дауна, трисомия по 21-й хромосоме - самая частая и наиболее хорошо изученная хромосомная болезнь. Описана в 1866 г. английским педиатром Л.Дауном. Частота рождения детей с синдромом Дауна составляет примерно 1:750 и не имеет какой-либо временной, этнической или географической разницы и родителей одинакового возраста. Цитогенетические варианты синдрома Дауна разнообразны. Основную долю составляют случаи полной трисомии 21 как следствие не расхождения хромосом в мейозе (кариотип – 47, XX(XY) +21). Наряду с этим известны случаи регулярной трисомии, связанной с транслокацией 21-й хромосомы на другую - 21, 22, 13, 14 или 15-ю хромосому. Почти 50% транслокационных форм наследуется от родителей носителей и 50 % - вновь возникшие мутации. Соотношение мальчиков и девочек среди новорожденных с синдромом Дауна составляет 1:1.
Пороки развития, нарушения постнатального развития нервной системы, иммунодефициты и другие отклонения. Многие симптомы заметны уже при рождении ребенка и дальнейшем проявляются еще более отчетливо. Из черепно-лицевых дизморфий отмечается монголоидный разрез глаз, круглое уплощенное лицо, плоская спинка носа, крупный язык, брахицефалия, деформированные ушные раковины. Так же характерны мышечная гипотония и разболтанность суставов. Часто диагностируются врожденный порок сердца, клинодактилия. Встречаются изменения дерматоглифики в виде четырехпальцевой, или "обезьяньей", складки на ладони, две кожные складки вместо трех на мизинце. Характерен низкий рост (на 20 см ниже среднего).
Диагноз синдрома Дауна ставится на основании клинически на основании сочетания ряда симптомов. Наиболее важные из которых: уплощение профиля лица (90 %), отсутствие сосательного рефлекса (85 %), избыток кожи на шее (80 %), монголоидный разрез глаз (80 %), мышечная гипотония (80 %), разболтанность суставов (80 %), диспластический таз (70 %), деформированные ушные раковины (40 %), клинодактилия мизинца (60 %), четырехпальцевая сгибательная складка (поперечная линия) на ладони (40 %).
Большое значение для диагностики имеет задержка умственного и физического развития ребенка. Задержка умственного развития может достигать степени имбицильности, а коэффициент умственного развития у разных детей широко варьируется (IQ от 25 до 75). Больные с синдромом Дауна часто болеют пневмониями, тяжело переносят детские инфекции. У них отмечается недостаток массы тела. Врожденные пороки внутренних органов и недостаточность иммунной системы часто приводят к летальному исходу в первые 5 лет жизни. Дифференциальная диагностика проводится с другими формами хромосомных аномалий и врожденным гипотиреозом. Цитогенетическое исследование показано и при подозрении на синдром Дауна и при клинически установленном диагнозе. В последнем случае это необходимо для прогноза здоровья будущих детей у родителей ребенка и их родственников. Лечебная помощь детям с синдромом Дауна многопланова и неспецифична. Врожденные пороки сердца устраняют оперативно. Постоянно проводится общеукрепляющая терапия, защита от действия вредных факторов внешней среды. Многие больные с трисомией 21 способны вести самостоятельную жизнь, овладевают несложными профессиями, создают семью.
С возрастом (в большей степени матери и в меньшей мере отца) вероятность рождения ребенка с данной патологией существенно возрастает, и в возрасте 45 лет составляет около 3 %.
Синдром Патау.
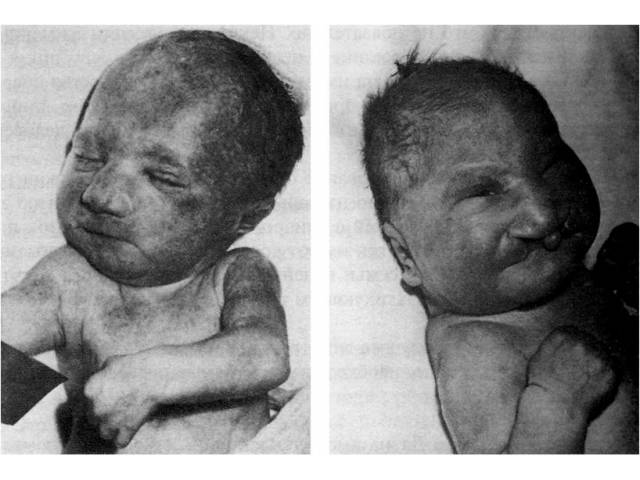
Трисомия по 13-й хромосоме, выделен в самостоятельную нозологическую форму в 1960 г., в результате генетического исследования у детей с врожденными пороками развития, американским генетиком К.Патау.
Обнаружены простые и транслокационные формы трисомии 13, однако клинически и патологоанатомически они неразличимы. Простая трисомия следствие не расхождения 13 пары хромосом в мейозе у одного из родителей. Частота синдрома Патау среди новорожденных составляет 1:6000. Соотношение полов при данной патологии близко 1:1.
Дети с синдромом Патау рождаются с истинной пренатальной гипоплазией. Их средняя масса при рождении составляет 2500 г, что почти на 900 г меньше средней массы детей при рождении. Продолжительность беременности практически не изменена, осложнением ее почти в половине случаев является многоводие.
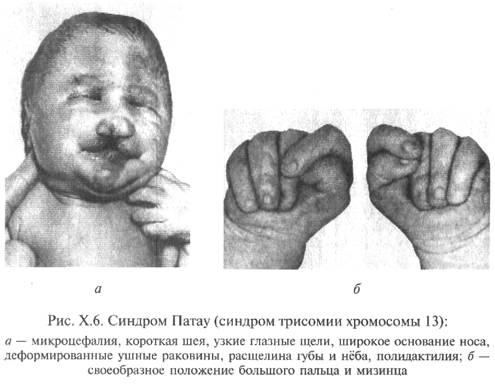
Фенотипические признаки синдрома настолько характерны, что позволяют практически сразу заподозрить это заболевание. Особенно обращают на себя внимание аномалии черепа и лица - микроцефалия, в ряде случаев отмечается выраженная тригоноцефалия, скошенный лоб, узкие глазные щели, \, запавшее переносье, низкорасположенные и деформированные ушные раковины. На коже головы имеются дефекты скальпа овальной или округлой формы, до 1 см в диаметре, дно таких дефектов представлено апоневротическим шлемом.
Наиболее характерными внешними пороками развития являются расщелина губы и нёба и полидактилия.
Врожденные пороки сердца отмечаются у 80% детей. Пороки пищеварительного тракта отмечаются у половины больных. Наиболее часто встречаются незавершенный поворот кишечника, Меккелев дивертикул, нарушение лобуляции печени, гетеротопия в поджелудочную железу ткани селезенки. Пороки развития почек наблюдаются в 60% случаев, наиболее характерным является поликистоз. Половые органы поражаются более чем в 50% случаев — у девочек удвоение матки и влагалиша, у мальчиков — гипоплазия полового члена и крипторхизм. Пороки развития органов зрения — анофтальмия, микрофтальмия, дисплазии сетчатки, колобома радужки, помутнение хрусталика — встречаются более чем у 70% больных. Центральная нервная система поражается в 100% случаев. Наиболее постоянны пороки переднего мозга.
Продолжительность жизни у детей с синдромом Патау резко снижена. На первом году жизни умирают 95% больных, причем 60-65% в перинатальном периоде. В возрасте старше 3 лет остаются в живых единицы. Все дети с синдромом Патау имеют тяжелую умственную отсталость (глубокая идиотия.
Лечебные мероприятия неспецифичны: общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. некоторые больные живут до нескольких лет.
Синдром Эдвардса.
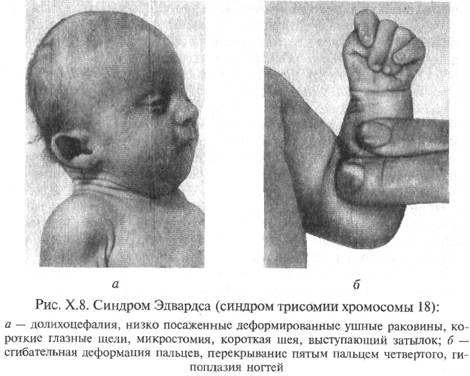
Синдром Эдвардса получил название по имени английского цитогенетика, впервые описавшего его хромосомную природу в 1960г. при обследовании ребенка с множественными пороками и аномалиями развития. Оказалось, что причиной синдрома практически во всех случаях является полная трисомия по 18-й хромосоме, возникающая в результате нерасхождения 18-й пары хромосом во время мейоза.
Частота синдрома среди новорожденных составляет 1:7000; девочки болеют примерно в три раза чаще мальчиков. Во время беременности отмечаются слабая двигательная активность плода, многоводие.
Больные с синдромом Эдвардса рождаются с низкой массой тела (в среднем 2200 г).
Фенотипические проявления: череп долихоцефалической формы, нижняя челюсть и отверстие рта маленькие, низкорасположенные, наружный слуховой проход сужен. Грудина короткая, грудная клетка – широкая. Характерное сгибательное положение пальцев кисти, выступающий затылок и другие микроаномалии.
При синдроме практически постоянны пороки сердца и крупных сосудов, часты пороки желудочно-кишечного тракта, пороки почек и половых органов.
Продолжительность жизни больных с синдромом Эдвардса резко снижена. На первом году жизни погибают 90% больных, к 3-летнему возрасту — более 95%. Причиной смерти являются пороки сердечно-сосудистой системы, кишечника или почек. Все выжившие больные имеют глубокую степень олигофрении (идиотию).
АНОМАЛИИ ЧИСЛА ПОЛОВЫХ ХРОМОСОМ
Это большая группа хромосомных болезней, представленная различным комбинациями дополнительных Х- или Y-хромосом, а в случаях мозаицизма – комбинациями различных клонов.
Изменение числа половых хромосом может возникать в результате нарушения расхождения как в первом, так и во втором делении мейоза. Нарушение расхождения в первом делении приводит к образованию аномальных гамет: у женщин — XX и 0 (в последнем случае яйцеклетка не содержит половых хромосом); у мужчин — XY и 0. При слиянии гамет во время оплодотворения возникают количественные нарушения половых хромосом.
1.Синдром трисомии Х. Частота синдрома трисомии X (47, XXX) составляет 1:1000-1:2000 новорожденных девочек. Впервые была описана в 1959 г. А.Джекобсом
Как правило, физическое и психическое развитие у больных с этим синдромом не имеет отклонений от нормы. Это объясняется тем, что у них активируются две Х-хромосомы, а одна продолжает функционировать, как у нормальных женщин. Изменения в кариотипе обнаруживаются случайно при обследовании. Умственное развитие также обычно нормально, иногда на нижних границах нормы. Лишь у некоторых женщин отмечаются нарушения со стороны репродуктивной функции (различные нарушения цикла, вторичная аменорея, ранняя менопауза).
При тетрасомиях X отмечаются высокий рост, телосложение по мужскому типу, эпикант, гипертелоризм, уплощенное переносье, высокое нёбо, аномальный рост зубов, деформированные и аномально расположенные ушные раковины, клинодактилиямизинцев, поперечная ладонная складка. У этих женщин описаны различные нарушения менструального цикла, бесплодие, преждевременный климакс.
Снижение интеллекта от пограничной умственной отсталости до рахчичных степеней олигофрении описано у двух третей больных. Среди женщин с полисемией X увеличена частота психических заболеваний (шизофрения, маниакально-депрессивный психоз, эпилепсия).
Синдром Клайнфельтера.
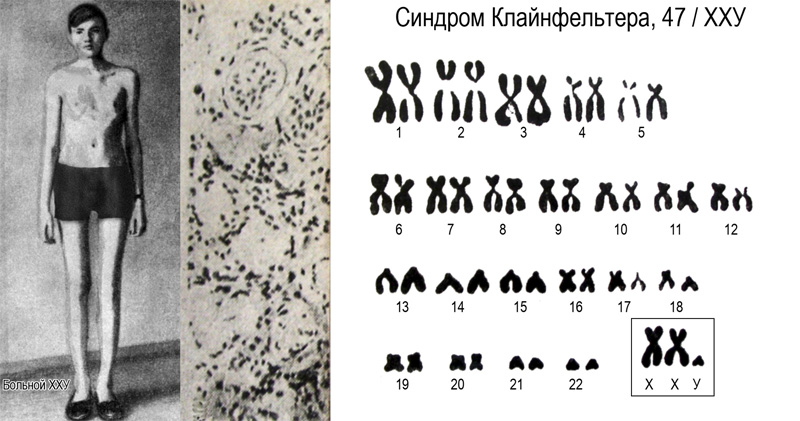
Впервые это синдром был описан в 1942 г. Г.Ф.Клайнефельтером. Синдром Клайнфельтера наблюдается у 1 из 500-700 новорожденных мальчиков; у 1-2,5% мужчин, страдающих олигофренией (чаще при неглубоком интеллектуальном снижении); у 10% мужчин, страдающих бесплодием.
В периоде новорожденности заподозрить этот синдром практически невозможно. Основные клинические проявления манифестируют в пубертатном периоде. Классическими проявлениями этого заболевания считаются высокий рост, евнухоидное телосложение, гинекомастия, но все эти симптомы одновременно встречаются лишь в половине случаев. Мужчины бесплодны.
У больных отмечается скудность или отсутствие оволосенения на лице, в подмышечных впадинах, на груди. На лобке оволосенение располагается по женскому типу. У этих пациентов достаточно часто отмечаются уплощенный затылок, гипертелоризм, эпикант, выступающие надбровные дуги, высокое небо, аномальный рост зубов. Умственная отсталость отмечается в 20-25% случаев. Степень умственной отсталости колеблется от пограничных состояний до дебильности различной тяжести.
Увеличение числа Х-хромосом (48, ХХХY, 49, ХХХХY) в кариотипе ведет к большей степени интеллектуального дефекта и более широкому спектру симптомов у пациентов.
3. Синдром дисомии по Y-хромосоме впервые описали А. А. Сандберг с соавторами в 1961 г., кариотип больных с этим заболеванием — 47, ХYY.
Частота этого синдрома среди новорожденных мальчиков составляет 1:840 и возрастает до 10 % у высокорослых мужчин (выше 200 см). У большинства больных отмечается ускорение темпов роста в детском возрасте. Средний рост у взрослых мужчин составляет 186см. В большинстве случаев по физическому и умственному развитию больные не отличаются от нормальных индивидов. Заметных отклонений в половой и в эндокринной сфере нет. В 30-40% случаев отмечаются определенные симптомы - грубые черты лица, выступающие надбровные дуги и переносица, увеличенная нижняя челюсть, высокое нёбо, аномальный рост зубов с дефектами зубной эмали, большие ушные раковины, деформация коленных, локтевых суставов. Интеллект или негрубо снижен, или в норме. Характерны эмоционально-волевые нарушения: агрессивность, взрывчатость, импульсивность. В то же время для этого синдрома характерны подражательность, повышенная внушаемость, причем больные наиболее легко усваивают негативные формы поведения.
Продолжительность жизни у таких больных не отличается от среднепопуляционной.