
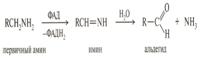
Примером может быть получение спиртов при окислении пищи. За счет молекулярного кислорода и его активных форм. осуществляется в in vivo.
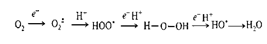
Перекись водорода может служить в организме гидроксилирующим агентом.
Избыток перекиси должен разлагаться с помощью каталазы на воду и кислород.
Окисление и восстановление алкенов можно представить следующими превращениями:



Окисление ароматических углеводородов
Бензол чрезвычайно тяжело окисляется даже в жестких условиях по схеме:
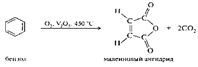
Способность к окислению заметно увеличивается от бензола к нафталину и далее к антрацену.
ЭД- заместители облегчают окисление ароматических соединений. ЭА – затрудняют окисление.
Ферментативное гидроксилирование ароматических соединений
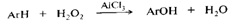


12. Альдегиды и кетоны относятся к карбонильным органическим соединениям.
Карбонильными соединениями называют органические вещества, в молекулах которых имеется группа >С=О (карбонил или оксогруппа). Общая формула карбонильных соединений: 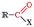 В зависимости от типа заместителя Х эти соединения подразделяют на: альдегиды (Х = Н); кетоны (Х = R, R'); карбоновые кислоты (Х = ОН) и их производные
В зависимости от типа заместителя Х эти соединения подразделяют на: альдегиды (Х = Н); кетоны (Х = R, R'); карбоновые кислоты (Х = ОН) и их производные
(Х = ОR, NH2, NHR, Hal и т.д.).
Альдегиды - органические соединения, в молекулах которых атом углерода карбонильной группы (карбонильный углерод) связан с атомом водорода. Общая формула: R–CН=O
Функциональная группа –СН=О называется альдегидной.
Кетоны - органические вещества, молекулы которых содержат карбонильную группу, соединенную с двумя углеводородными радикалами. Общие формулы: R2C=O, R–CO–R' СН3—СО—СН3 и называется диметилкетоном
Окисление и восстановление альдегидов и кетонов
Один из наиболее легко окисляющийся классов органических соединений 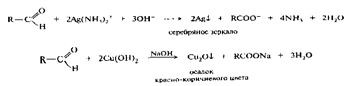
2Н2С = О + Н2О СН3ОН + НСООН особенно легко протекает на свету
13. Карбоновые кислоты – органические соединения, содержащие одну или несколько карбоксильных групп -СООН.
Карбоксильная группа содержит две функциональные группы – карбонил >С=О и гидроксил -OH, непосредственно связанные друг с другом: 
По числу карбоксильных групп различают одно-, двух-, многоосновные кислоты.
К производным карбоновых кислот относятся соединения общей формулы:

H-COOH муравьиная кислота метановая кислота
С4H9-СООН валериановая пентановая
Первые четыре члена ряда кислот смешиваются с водой, пятый член – частично растворим, высшие кислоты практически не растворимы в воде. Простейшие ароматические кислоты не растворимы в воде. Температура кипения кислот выше чем у спиртов. Это связано с тем, что карбоновые кислоты связаны не одной, а двумя водородными связями 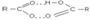
Окисление парафиновых углеводородов воздухом или техническим кислородом.

При окислении получается смесь кислот. В качестве катализатора используют металлы, их соли, оксиды. Низшие углеводороды окисляются в паровой фазе, высшие углеводороды – в жидкой фазе.
Синтезы уксусной кислоты:

В карбоновых кислотах имеются пять реакционных центров, где возможно протекание химических реакций:
1) кислотный на связи О – Н
2) протоноакцепторный на атоме кислорода карбоксильной группы -С(О)ОН.
3) диссоциативный по связи -С(О) – ОН
4) диссоциативный по связи - С(О)ОН
5) замещение атома водорода в алкильной (арильной) группе R.
14. Кофермент А - кофермент ацетилирования; один из важнейших коферментов, принимающий участие в реакциях переноса ацильных групп при синтезе и окислении жирных кислот и окислении пирувата вцикле лимонной кислоты. Молекула КоА состоит из остатка адениловой кислоты (1), связанной пирофосфатной группой (2) с остатком пантоевой кислоты(3), которая в свою очередь связанна пептидной связью с аминокислотой β-аланином (4) (эти две группы представляют собой остаток пантотеновой кислоты), соединённой пептидной связью с остатком β-меркаптоэтаноламина. C21H36N7O16P3S

Кофермент А синтезируется в пять этапов из пантотеновой кислоты. С КоА связан ряд биохимических реакций, лежащих в основе окисления и синтеза жирных кислот, биосинтеза жиров, окислительных превращений продуктов распада углеводов. Во всех случаях КоА действует в качестве промежуточного звена, связывающего и переносящего кислотные остатки на другие вещества. При этом кислотные остатки в составе соединения с КоА подвергаются тем или иным превращениям, либо передаются без изменений на определённые метаболиты.
15. Двухосновные карбоновые кислоты (или дикарбоновые кислоты) — это карбоновые кислоты, содержащие две карбоксильные группы —COOH, с общей формулой HOOC—R—COOH, где R — любой двухвалентный органический радикал. Дикарбоновые кислоты проявляют те же химические свойства, что и монокарбоновые — эти свойства обусловлены наличием карбоксильной группы:
диссоциация в водных растворах: первая стадия (Ka1):
НOOC—Х—СООН → НOOC—Х—СОО− + Н+
Дикарбоновые кислоты — более сильные кислоты по первой стадии диссоциации, чем соответствующие монокарбоновые: во-первых, из-за статистического фактора (две карбоксильных группы в молекуле), во-вторых, из-за взаимного влияния этих групп (если они находятся недалеко или связаны цепью кратных связей);
вторая стадия (Ka2):
НOOC—Х—СОО− → −OОC—Х—СОО− + Н+
На второй стадии эти кислоты становятся более слабыми, чем монокарбоновые кислоты (исключение — щавелевая кислота). Отделение катиона водорода второй карбоксильной группы происходит труднее, чем первой, так как требуется больше энергии, чтобы отделить H+ от аниона с зарядом −2, чем при отделении от аниона с зарядом −1;
образование солей: в отличие от монокарбоновых кислот, дикарбоновые способны образовывать кислые соли; образование галогенангидридов.
В то же время есть существенные различия, обусловленные наличием второй карбоксильной группы: склонность к образованию хелатов; образование некоторыми кислотами циклических ангидридов; способность образовывать полимеры в реакции с другими полифункциональными соединениями.
16. Поли- и гетерофункциональность, как причина появления специфических свойств у гидрокси-, амино- и оксокислот.
Наличие в молекуле нескольких одинаковых или разных функциональных групп составляет характерную черту биологически важных органических соединений. В молекуле может быть две и более гидроксильных групп, аминогрупп, карбоксильных групп. Например:
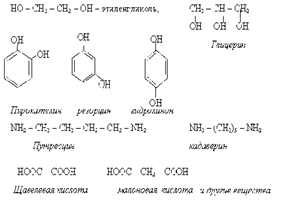
Важную группу веществ участников жизнедеятельности составляют гетерофункциональные соединения, имеющие попарное сочетание разных функциональных групп. Например:
В алифатических соединениях все приведённые функциональные группы проявляют ЭА характер. За счёт влияния друг на друга у них взаимно усиливается реакционная способность. Например, в оксокислотах электрофильность усиливается каждого из двух карбонильных атомов углерода под влиянием -J другой функциональной группы, что ведёт к более легкому восприятию атаки нуклеофильными реагентами.
 Поскольку I эффект затухает через 3–4 связи, то важным обстоятельством является близость расположения функциональных групп в углеводородной цепи. Гетерофункциональные группы могут находится у одного и того же атома углерода (альфа – расположение), или у разных атомов углерода как соседних(бета расположение), так и более удалённых друг от друга (гамма, дельта, эпсилон) расположения.Каждая гетерофункциональная группа сохраняет собственную реакционную способность, точнее гетерофункциональные соединения вступают как бы в «двойное» число химических реакций. При достаточном близком взаимном расположении гетерофункциональных групп происходит взаимное усиление реакционной способности каждой из них.
Поскольку I эффект затухает через 3–4 связи, то важным обстоятельством является близость расположения функциональных групп в углеводородной цепи. Гетерофункциональные группы могут находится у одного и того же атома углерода (альфа – расположение), или у разных атомов углерода как соседних(бета расположение), так и более удалённых друг от друга (гамма, дельта, эпсилон) расположения.Каждая гетерофункциональная группа сохраняет собственную реакционную способность, точнее гетерофункциональные соединения вступают как бы в «двойное» число химических реакций. При достаточном близком взаимном расположении гетерофункциональных групп происходит взаимное усиление реакционной способности каждой из них.
При одновременном присутствии в молекуле кислотной и основной групп, соединение становятся амфотерным.
Например: аминокислоты. 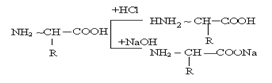
19. Гетероциклы с 2-мя атомами азота
Важнейшими из которых являются пиразол, пиримидин, имидазол. Пятичленые гетероциклы с 2 гетероатомами: пиразол и имидазол являются структурными изомерами и близки по химическим свойствам.
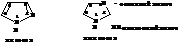
Образуют межмолекулярные водородные связи. По этой причине имидазол имеет аномально высокую температуру кипения. Имидазол участвует в кислотно-основном катализе и в реакциях SN
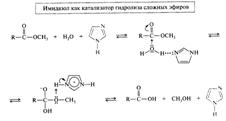
Следствием такой ассоциации является быстрый водородный обмен, который в свою очередь является причиной прототропной таутомерии у производных этих гетероциклов, а также конденсированного гетероцикла пурина. Поэтому, например, у имидазола положение 1 и 3, а у пурина положения 7 и 9 равноценны. Замещение в NH-группе водорода, устраняет возможность таутомерии.

Имидазольное ядро является структурным фрагментом гистидина, которая при декарбоксилировании образует биогенный амин– гистамин, который отвечает за аллергические реакции организма. 
Приразол и его производные в природе не встречаются, но на его основе синтезированы известные анальгетики - антипирин, амидопирин, анальгин.
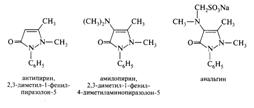
21.


Особенно важны гидрокси- и аминопроизводные пиримидина — урацил (2,4-дигидроксипиримидин), тимин (2,4-дигидрокси-5-метилпиримидин) и цитозин (4-амино-2-одроксипиримидин) — компоненты нуклеиновых кислот. Для урацила и тимина характерна лактим-лактамная таутомерия.


Обычно лактамная форма в равновесии преобладает. В лактимной форме, т. е. гидроксиформе, пиримидиновое ядро обладает ароматичностью. Однако и в лактамной форме ароматичность не нарушается, так как ароматическая система образуется за счет участия в сопряжении неподеленной пары электронов амидного атома азота. Разрыв сопряжения в кольце отсутствует. В случае аминопроизводных пиримидина может иметь место также енамино-иминная таутомерия. В частности, для цитозина помимо лактамной и лактимной форм в принципе возможны и иминные формы, но преобладающими являются енаминные формы.
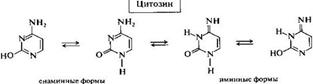
Урацил, тимин, цитозин — твердые высокоплавкие вещества, растворимые в воде и нерастворимые в неполярных органических растворителях. Для них характерно наличие прочных межмолекулярных водородных связей.

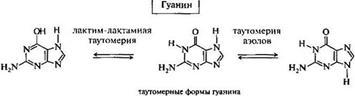
Наиболее важны 6-аминопурин, или аденин, и 2-амино-б-гидроксипурин, или гуанин, являющиеся обязательными компонентами нуклеиновых кислот. Аденин также входит в состав некоторых коферментов. Для аденина возможна азольная прототропная таутомерия за счет миграции атома водорода между N-7 и N-9.

Для гуанина возможна также лактим-лактамная таутомерия.
24. α-аминокислоты содержат амино- и карбоксильную группу у одного и того же атома С и отличаются строением радикала R. Аминокислоты классифицируют как: Ароматические,алифатич.,гетероциклические Заменимые и незаменимые
Полярные и не полярные
Кислые, основные и нейтральные
Гидрофобные и гидрофильные
Ионогенные и неионогенные
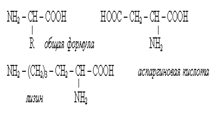
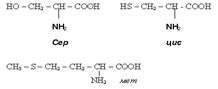
В природных α-аминокислотах кроме одной амино- и карбоксильной группы содержатся и дополнительные функциональные группы, еще по одной NH2 или СООН, а также ОН, SH, CH3S, гетероциклические фрагменты. В природе найдено 150 аминокислот, в состав белков животного происхождения входит около 20.Н-р:
Номенклатура и стереоизомерия α - аминокислот
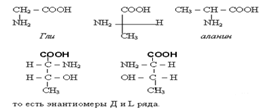
Чаще используются тривиальные названия α-аминокислот. В записях названий сложных соединений, построенных из остатков аминокислот (пептиды, белки) применяют 3-х буквенные обозначения (обычно три первые буквы). Примеры будут даны по ходу изложения. Кроме глицина все аминокислоты имеют ассиметричный атом С,т.е имеют D и L изомеры.Изолейцин,треонин,цистин,4-гидроксипролин имеют по два хиральных атома углерода и имеют по четыре стереоизомера.
В белках живых организмов представлены L- аминокислоты, в белках микроорганизмов встречаются аминокислоты и Д- ряда. Д-аминокислоты нашим организмом не усваиваются.
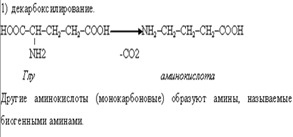
Биосинтез α-аминокислот осуществляется из α-кетокислот,которые являются продуктами метаболизма углеводов.
Возможно 2 пути превращение кетонокислот в аминокислоты.
1) восстановительное аминирование с участием НАДН.

2) трансаминировние (переаминирование), когда источником группы NH2 для кетокислоты является другая аминокислота.
Реакция протекает в присутствии кофермента пиридоксальфосфата
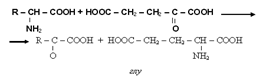
25. Химические свойства. Аминокислоты амфотерные соединения, так как содержат и кислотный и основной центры, следовательно, в среде, близкой к нейтральной, они существуют в виде внутренних солей (биполярных ионов). Аминокислотам присущи все реакции как карбоновых кислот, так и аминов. Реакции карбоксильной группы – образование функциональных производных (сложных эфиров, амидов, ангидридов и др.). Реакции NH2-, образование солей с сильными кислотами, имидов. Одна из реакций аминов взаимодействие с HNO2 – метод количественного определения аминокислот (метод Ван-Слейка).
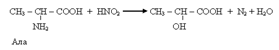
26. Ряд специфических реакций обусловлен наличием СООН и NH2 у одного и того же атома С. В основном – это реакции, протекающие в организме, то есть ферментативные реакции (хотя их можно провести и вне организма). К таким реакциям относятся:
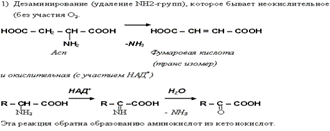
В организме осуществляется гидроксилирование некоторых аминокислот, которые не имеют аналогов в химии IN VITRO.
Например гидроксилирование фенилаланина с образованием тирозина и 4-гидроксипролина из пролина.

27. Строение пептидов

Пептиды содержат от 50 до 100 остатков аминокислот, белки более 100. Граница деления условна. Последовательность остатков аминокислот в пептидах и белках характеризует их первичную структуру.
Химические свойства пептидов и белков вытекают из их амидной природы: они способны гидролизоваться в кислой среде и в щелочной среде, в организме гидролиз осуществляется под действием ферментов – протеиназ.
Гидролиз пептидов даёт сведения об аминокислотах, входящих в состав пептида. При этом необходимо учитывать, что некоторые аминокислоты частично или полностью разрушаются в условиях жесткого кислотного гидролиза. При гидролизе пептиды полностью теряется информация о порядке соединения аминокислот в цепи, то есть о первичной структуре.

кроме первичной структуры существует и вторичная структура.Пространственное расположение полипептидной цепи во вторичной структуре фиксируется водородными связями. Так отдельные участки цепи образуют α-спираль, как это было показано Полингом или β-складчатый лист.
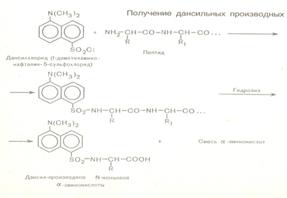
28. Методы определения аминокислотной последовательности (установление первичной структуры) хорошо разработаны. Это метод динитрофенилирования, метод Эдмана, дансильный метод.
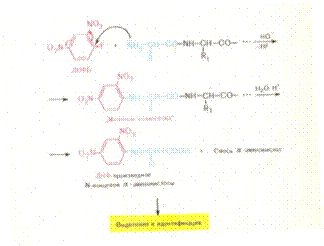

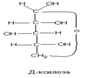
29. Моносахариды классифицируются по:
а) характеру карбонильной группы на альдозы и кетозы.
б) по числу С в цепи на тетрозы,пентозы,гексозы. Следовательно, с учетом обоих признаков моносахариды делятся на альдопентозы, альдогексозы, кетопентозы, кетогексозы. Каждый моносахарид содержит несколько хиральных атомов С, и для них возможно существование стереоизомеров, количество которых определяется по формуле 2 в степени n.
Моносахариды также делят на моносахариды Д ряда и L ряда.
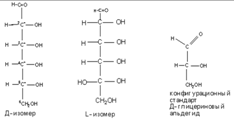
Конкретные названия моносахаридов определяются конфигурацией всех ассиметричных атомов углерода. Отнесение к Д или L рядам проведется по конфигурации наиболее удаленного хирального атома С. В природе чаще встречаются моносахариды Д – ряда, хотя встречаются и L – ряда.
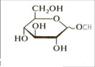
Циклическое строение. Выше приведенным строением моносахаридов нельзя было объяснить все химические свойства моносахаридов. Это навело на мысль об ином, а именно, о циклическом строении моносахаридов. Впервые эта идея была высказана русским химиком А.А. Колли (1870) и развита позднее Б. Толленсом (1883 г.).
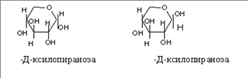
Атом С-1 становится в результате циклизации ассиметричным,следовательно образовавшаяся при циклизации ОН может находиться как над- так и под плоскостью цикла. Эта ОН группа называется полуацетальной (гликозидной) оба стереоизомера не являются зеркальным отражением друг друга, поэтому они диастереоизомеры и называются аномерами. Если ОН находится под плоскостью цикла, то это α- аномер, если над β-аномер.
Циклическая полуацетальная форма может образоваться и за счет ОН группы у 4-го С,с образованием пятичленного фуранозного цикла,а при 5-ом пиранозного шестичленного цикла.
Рассмотрим это на примере образования фуранозных форм Д-рибозы
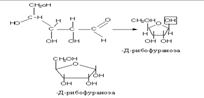
Приведенные циклические формулы называются формулами Хеуорса (1928г.). В формулах циклический атом кислорода изображают в правом дальнем углу, атом С-1-справа. Группа ОН, которые находились в открытой форме справа располагаются над плоскостью цикла, а те, что в открытой форме были слева над плоскостью.

30. Цикло-оксо-таутомерия.

Несмотря на незначительное содержание в открытой форме глюкозы она вступает в реакции характерные для карбонильной группы (-СОН).
Соотношение таутомеров различное для разных моносахаридов. Более устойчивы пиранозные циклы. Так для D-глюкозы открытой формы лишь 0,003-0,03%, но через нее происходит превращение циклических форм друг в друга.
Например α-глюкоза в воде постепенно за несколько часов дает смесь α и β-пиранозных циклов в соотношении 1:2. Из β-D – глюкопиранозы образуется смесь того же состава. Если вам удастся найти в индивидуальном виде открытую форму D-глюкозы, то и она со временем дает смесь α и β-пиранозных форм в соотношении 1:2.
Равновесие между α и β - аномерами в формулах Хеуорса изображены волнистой линией, как это показано на примере Д-глюкуроновой кислоты.
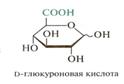
Конформационным строением объясняется соотношение α и β- пиранозных форм D-глюкозы в растворе. Таутомерное равновесие сдвинуто в сторону β-аномера, т.к.наиболее устойчивы аномеры с экваториальным расположением заместителей.

В α-D-глюкозе СН2ОН и все ОН кроме полуацетального ОН занимают экваториальное положение. β-экваториальное положение полуацетального гидроксила в конформации моносахаридов, особенно с учетом конфигурации аномерного центра (α или β) очень важна для пространственного строения полисахаридов.

31.. Химические свойства. Моносахариды весьма реакционноспособные соединения вследствие их поли- и гетерофункциональности. Свойства моносахаридов можно рассматривать как свойства:
Полуацетального гидроксила(образование О- и N-гликозидов)
Cпиртовых гидроксилов(образование простых и сложных эфиров и фосфатов)
Свойства карбонильной группы(окисление и восстановление)
Свойства С-Н кислотного центра(эпимиризация и изомеризация)
Аналогично взаимодействиям карбонильных соединений со спиртами в присутствии Н+ и без Н2О моносахариды образуют полуацетали называющиеся гликозидами. Например:
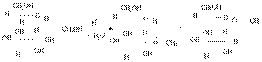
Метил- α-и-β-D-глюкопиранозиды
Смесь можно изобразить одной формулой
Это знакомая реакция нуклеофильного замещения. Механизм ее аналогичен образованию обычных ацеталей. Роль Н+ заключется в превращении ОН- из плохо уходящей в хорошо уходящую группу Н2О.
Но, а как в организме образуется гликозиды в присутствии сильных кислот да еще в безводной среде?. В iп vivo для этой цепи используются фосфаты или чаще нуклеозиддифосфат сахара.

В организме моносахариды образуют гликозиды за счет спиртовых ОН групп самих же моносахаридов и полуацетального гидроксила. При таких взаимодействиях образуюся олиго- и полисахариды.
Кроме того, в организме происходит образование гликозидов D-глюкуроновой кислоты с некоторыми токсинами, и фенольными соединениями. Эти гликозиды выводятся из организма с мочой и осуществляется тем самым детоксикация.
Гликозидам родственны их азотсодержащие соединения, которые называются N-гликозидами.N- гликозиды являются компонентами РНК и ДНК.
Важнейшее свойство гликозидов это способность подвергаться гидролизу в кислой среде и достаточная устойчивость к гидролизу в щелочной среде.
Гидролиз гликозидов – реакция обратная их образованию.

32. Восстановление. При восстановлении моносахаридов (их альдегидной или кетонной групп) образуются многоатомные спирты (полиолы) называемые альдитами. Эти кристаллические легко растворимые в воде вещества обладают сладким вкусом и часто используются как заменители сахара при сахарном диабете (ксилит, сорбит).
Восстановление моносахаридов проводят водородом в присутствии металлических катализаторов (палладий, никель). Шестиатомные спирты — глюцит (сорбит), дульцит и маннит — получаются при восстановлении соответственно глюкозы, галактозы и маннозы. Восстановление глюкозы в сорбит является одной из стадий промышленного синтеза аскорбиновой кислоты.
окисление в щелочной среде. Альдозы в связи с наличием альдегидной группы способны восстанавливать в щелочкой среде катионы металлов (серебра, меди). Такие реакции возможны за счет таутомерного перехода в альдегидную форму, например реакция серебряного зеркала с реактивом Толленса. Кетозы тоже способны восстанавливать катионы металлов, так как они в щелочной среде изомеризуются в альдозы. Моносахариды в щелочной среде неустойчивы, поэтому при их окислении получается смесь продуктов.
Окисление в нейтральной и кислой средах. Окисление альдоз без деструкции (разрушения) углеродного скелета проводят в нейтральной или кислой среде и получают различные кислоты. С помощью сильного окислителя — разбавленной азотной кислоты — концевые группы альдоз (альдегидная и первичноспиртовая) одновременно окисляются в карбоксильные группы, образуя альдаровые кислоты (называемые также сахарными)
37. Нуклеоти́ды — фосфорные эфиры нуклеозидов, нуклеозидфосфаты. Свободные нуклеотиды, в частности АТФ, цАМФ, АДФ, играют важную роль в энергетических и информационных внутриклеточных процессах, а также являются составляющими частями нуклеиновых кислот и многих коферментов. Нуклеотиды являются сложными эфирами нуклеозидов и фосфорных кислот. Нуклеозиды, в свою очередь, являются N-гликозидами, содержащими гетероциклический фрагмент, связанный через атом азота с C-1 атомом остатка сахара. Большинство нуклеотидов являются моноэфирами ортофосфорной кислоты, однако известны и диэфиры нуклеотидов, в которых этерифицированы два гидроксильных остатка
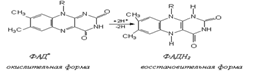
38. Никотинамиднуклеотиды. Наиболее важный представитель это группы соединений никотинамидадениндинуклеотид (НАДН) или его фосфат НАДФН (кофермент дегидрогеназ), то есть являются участниками окислительно-восстановительных реакций. В соответствии с этим, могут находится в окисленной и восстановленной формах.
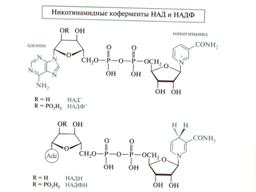

Причем реакции с участием НАДН и НАДФН протекают стереоселективно, т.е с образованием одного из стереоизомеров. Н-р

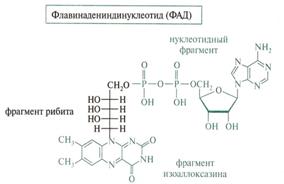
Флавинадениндинуклеотид -кофермент окислительно-восстановительных процессов с переносом двух электронов,которая является активной формой витамина В2(рибофловина).
39. АТФ, англ. АТР) — нуклеотид, играет исключительно важную роль в обмене энергии и веществ в организмах; в первую очередь соединение известно как универсальный источник энергии для всех биохимических процессов, протекающих в живых системах. Главная роль АТФ в организме связана с обеспечением энергией многочисленных биохимических реакций. Являясь носителем двух высокоэнергетических связей, АТФ служит непосредственным источником энергии для множества энергозатратных биохимических и физиологических процессов. Всё это реакции синтеза сложных веществ в организме: осуществление активного переноса молекул черезбиологические мембраны, в том числе и для создания трансмембранного электрического потенциала; осуществления мышечного сокращения.
Помимо энергетической АТФ выполняет в организме ещё ряд других не менее важных функций: Вместе с другими нуклеозидтрифосфатами АТФ является исходным продуктом при синтезе нуклеиновых кислот. Кроме того, АТФ отводится важное место в регуляции множества биохимических процессов. Являясь аллостерическим эффектором ряда ферментов, АТФ, присоединяясь к их регуляторным центрам, усиливает или подавляет их активность. АТФ является также непосредственным предшественником синтеза циклического аденозинмонофосфата — вторичного посредника передачи в клетку гормонального сигнала. Также известна роль АТФ в качестве медиатора в синапсах.
АТФ синтезируется при энергетическом обмене: вещества распадаются (окисляются), при этом выделяется энергия, которая запасается в макроэргических связях:
В организме АТФ синтезируется путём фосфорилирования АДФ:
АДФ + H3PO4 + энергия → АТФ + H2O.
40.
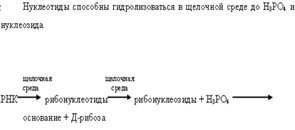
 Химический гидролиз ДНК осложнен из-за побочных процессов, более предпочтительны ферментативный гидролиз под действием нуклеаз (змеиный яд).
Химический гидролиз ДНК осложнен из-за побочных процессов, более предпочтительны ферментативный гидролиз под действием нуклеаз (змеиный яд).
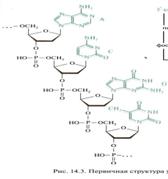
Понятие первичной структуры. НК – это нуклеотидный состав и нуклеотидная последовательность. Наиболее полно исследованы т-РНК, так как они имеют относительно низкую молекулярную массу.
Первичная структура нуклеиновых кислот – это нуклеотидный состав и определенная последовательность нуклеотидных звеньев в полимерной цепи.
41. Наиболее распространённой формой вторичной структуры ДНК является двойная спираль. Эта структура образуется из двух взаимно комплементарных антипараллельных полидезоксирибонуклеотидных цепей, закрученных относительно друг друга и общей оси в правую спираль[5]. При этом азотистые основания обращены внутрь двойной спирали, а сахарофосфатный остов — наружу. Впервые эту структуру описали Джеймс Уотсон и Френсис Крик в 1953 году[6].
В формировании вторичной структуры ДНК участвуют следующие типы взаимодействий:
· водородные связи между комплементарными основаниями (две между аденином и тимином, три — между гуанином и цитозином);
· стэкинг-взаимодействия;
· электростатические взаимодействия;
· Ван-дер-Ваальсовы взаимодействия.
В зависимости от внешних условий параметры двойной спирали ДНК могут меняться, причём иногда существенно. Правоспиральные ДНК со случайной нуклеотидной последовательностью можно грубо разделить на два семейства — А и В, главное отличие между которыми — конформация дезоксирибозы. К В-семейству также относятся С- и D-формы ДНК[7]. Нативная ДНК в клетке находится в В-форме. Важнейшие характеристики А- и В-форм ДНК приведены в таблице
конфигурация двойной спирали ДНК сильно меняется в зависимости от количественного содержания воды и ионной силыокружающей среды (формула)ДНК-АГЦТ
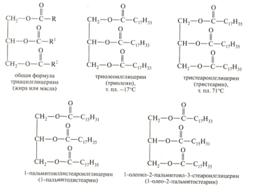
42. Жиры и масла (растительные жиры) представляют собой эфиры глицерина и высших жирных кислот, то есть это триацилглицерины. Простые липиды триацилглицерины. Простые: триацилглицирины содержат остатки одинаковых кислот, а смешанные – остатки различных кислот. Твердые триацилглицерины содержат остатки насыщенных кислот, а жидкие – ненасыщенные карбоновые кислоты.
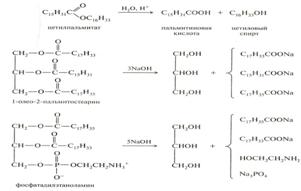
Химические свойства. Гидролиз протекает как в кислой, так и щелочной средах (омыление) и представляет собой обычную реакцию гидролиза сложного эфира. Гидролиз протекает ступенчато и продуктами полного гидролиза, являются глицерин и смесь высших жирных кислот.
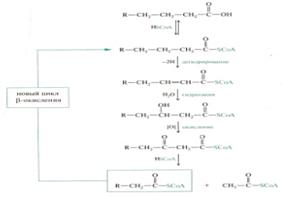
43. Бета – окисление. Окислению жиров предшествует гидролиз, а затем насыщенные карбоновые кислоты окисляются с расщеплением углеводородного скелета. При окислении жиров выделяется энергия 37,7 кДж/мольСвободная жирная кислота независимо от длины углеводородной цепи является метаболически инертной и не может подвергаться никаким биохимическим превращениям, в том числе окислению, пока не будет активирована. Свободная жирная кислота независимо от длины углеводородной цепи является метаболически инертной и не может подвергаться никаким биохимическим превращениям, в том числе окислению, пока не будет активирована.

В результате реакции образуется ацил-КоА, являющийся активной формой жирной кислоты. Ацетил-КоА может вступать в цитратный цикл и окисляться до СО2 и Н2О, а укороченный ацильный остаток будет вовлекаться в следующий цикл β-окисления. Каждый цикл включает четыре последовательные реакции, в результате которых жирная кислота укорачивается на два углеродных атома, которые отщепляются в виде ацетил-КоА. Ацетил-КоА может вступать в цитратный цикл и окисляться до СО2 и Н2О, а укороченный ацильный остаток будет вовлекаться в следующий цикл β-окисления.
44. Фосфолипиды - биологически активные вещества. Они входят в структурные клеточные мембраны и участвуют в транспорте жира в организме. B молекуле фосфолипидов глицерин этерифицирован ненасыщенными жирными кислотами и фосфорной кислотой, которая соединена с азотистым основанием. Из фосфолипидов в продуктах питания наиболее широко представлен лецитин, в молекуле которого две гидроксильные группы глицерина этерифицированы полиненасыщенными жирными кислотами, а третья гидроксильная группа глицерина соединена с фосфорной кислотой, в свою очередь связанной с аминоспиртом холином.
K фосфолипидам относится кефалин и сфингомиелин, обладающие действием, аналогичным действию лецитина.
B наибольшем количестве фосфолипиды представлены в нервной ткани и ткани мозга, сердца, печени и др. Фосфолипиды синтезируются в организме (печень, почки).
Лецитин - важный фактор регулирования холестеринового обмена. Он предотвращает накопление избыточных количеств холестерина в организме, способствует его расщеплению и выведению из организма. Фосфолипиды — сложные липиды, в которых содержатся жирные кислоты, фосфорная кислота и дополнительная группа атомов, во многих случаях содержащая азот. Они есть во всех живых клетках. Содержатся в нервной ткани, участвуют в транспорте жиров, жирных кислот и холестерина.
Фосфолипиды входят в состав всех клеточных мембран. Между плазмой и эритроцитами происходит обмен фосфолипидами, которые играют важнейшую роль, поддерживая в растворимом состоянии неполярные липиды. Наиболее распространенная группа Фосфолипидов — фосфоглицериды, также к фосфолипидам относятся фосфосфинголипиды и фосфоинозитиды.
Фосфолипиды — амфифильные вещества. Они состоят из полярной «головки», в состав которой входит глицерин или другой многоатомный спирт, отрицательно заряженный остаток фосфорной кислоты и часто несущая положительный заряд группа атомов, и двух неполярных «хвостов» из остатков жирных кислот. Главная особенность фосфолипидов состоит в том, что «головка» у них гидрофильна, а «хвосты» гидрофобны. Это позволяет при нахождении в толще водной среды образовывать бислой — двойной слой фосфолипидных молекул, где гидрофильные головы с обеих сторон соприкасаются с водой, а гидрофобные хвосты упрятаны внутрь бислоя и тем самым защищены от контакта с водой.
Это определяет многие физические и химические свойства фосфолипидов, например, способность формировать липосомы и биологические мембраны (липидный бислой). Химическая структура полярной «головки» определяет суммарный электрический заряд и ионное состояние фосфолипида. «Хвосты» контактируют с липидным окружением, а «головки» — с водным, так как неполярные жирные хвосты не могут соприкасаться с водой.
Лецитины — сложные эфиры аминоспирта холина и диглицеридфосфорных кислот; являются важнейшими представителями фосфолипидов. При расщеплении лецитинов образуются высшие жирные кислоты (пальмитиновая, стеариновая, олеиновая и арахидоновая), глицеро-фосфорная кислота и холин.
Лецитин — необходимое для организма вещество. Лецитин является основополагающим химическим веществом для формированния межклеточного пространства, нормального функционирования нервной системы, нормальной рабочей деятельности мозговых клеток, служит одним из основных материалов печени. Лецитин необходим организму как строительный материал для обновления поврежденных клеток. Лецитин это также основное транспортное средство для доставки питательных веществ, витаминов и лекарств к клеткам. Естественным источником лецитина являются продукты с высоким содержанием жира.
Кефалины), природные соединения из группы сложных липидов. Широко рас