при облучении мономера УФ-светом молекулы М + hv =М*=R1* + R2*
Однако прямое облучение мономера малоэффективно,поскольку кварцевое стекло не пропускает уф-свет в области, в соответствующей его поглощению мономером, или пропускает его в незначительной степени.
В том случае, когда мономер не поглощает прошедший свет, необходимо использовать фотосенсибилизатор (Z) соединение, передающее энергию возбуждения другим молекулам:
Z + hv =Z*, Z* + М = Z + М* = R1* + R2*+ Z.
Применение в качестве фотосенсибилизаторов красителей позволяет использовать для фотоинициирования видимую область света.
В практических целях фотополимеризация обычно проводится в присутствии фотоинициаторов - веществ, распадающихся в требуемой области УФ-спектра с достаточно высоким квантовым выходом. В качестве фотоинициаторов могут быть использованы некоторые термические инициаторы, например, пероксиды или азосоединения; а также другие соединения. Наиболее эффективными фотоинициаторами являются ароматические кетоны и их производные, благодаря достаточно широкой области поглощения УФ-спектра и высокому квантовому выходу радикалов. Считается, что ароматические кетоны претерпевают фотохимическое превращение по двум направлениям:
 последнее из которых реализуется лишь в присут. доноров водорода. В промышленности используют (1) бензоил, (2) бензилкеталь
последнее из которых реализуется лишь в присут. доноров водорода. В промышленности используют (1) бензоил, (2) бензилкеталь
 Фотополимеризация используется для нанесения полимерныхпокрытий непрерывным способом на металл, дерево, керамику, световоды, в стоматологии для отверждения композиций зубных пломб. В фотолитографии, с помощью которой изготавливают большие интегральные схемы в микроэлектронике, а также печатные платы (матрицы) в современной технологии фотонабора, позволяющей исключить использование свинца.
Фотополимеризация используется для нанесения полимерныхпокрытий непрерывным способом на металл, дерево, керамику, световоды, в стоматологии для отверждения композиций зубных пломб. В фотолитографии, с помощью которой изготавливают большие интегральные схемы в микроэлектронике, а также печатные платы (матрицы) в современной технологии фотонабора, позволяющей исключить использование свинца.
Существенным недостатком фотоинициирования является быстрое падение его эффективности с увеличением толщины облучаемого слоя вследствие поглощения излучения. По этой причине фотохимическое инициирование эффективно при возбуждении полимеризации в достаточно тонких слоях, порядка нескольких миллиметров.
Радиохимическое инициирование. Излучение радиоактивных источников Со60, а также различного рода ускорителей включает набор частиц таких как а-частицы, нейтроны, электроны и жесткое электромагнитное излучение. В отличие от фотоизлучения радиоактивное является ионизирующим и обладает гораздо большей проникающей способностью, что объясняет большей энергией его частиц.
Ионизация облучаемого вещества является следствием выбивания электронов из его молекул, например мономера, частицами высокой энергии:

 Радикалы, способные инициировать полимеризацию, возникают в результате дальнейших превращений в системе с участием возбужденных ионов, ионрадикалов и электронов, например:
Радикалы, способные инициировать полимеризацию, возникают в результате дальнейших превращений в системе с участием возбужденных ионов, ионрадикалов и электронов, например:
Наличие в облучаемом мономере свободных радикалов и ионов представляет возможность развития как радик. так и ионной полимеризации.
Термическое инициирование. Имеется очень мало примеров термического инициирования полимеризации. К ним относятся спонтанная полимеризация стирол а_и винилпиридинов. Считается, что механизм возникновения свободных радикалов при термическом инициировании является бимолекулярным, но достаточно надежно он выявлен лишь по отношению к стиролу. Первой стадией реакции является образование аддукта Дильса-Альдера из двух молекул стирола:
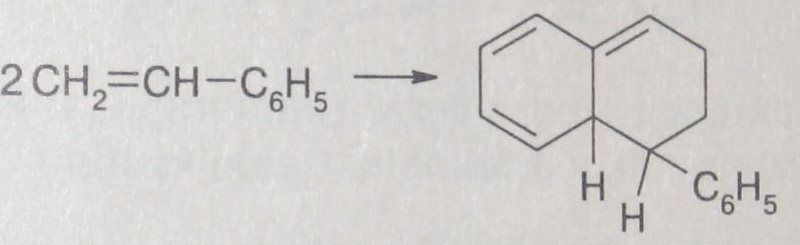
На второй стадии имеет место перенос атома водорода от аддукта к молекуле стирола, что и приводит к возникновению радикалов, способных инициировать полимеризацию:

В большинстве других случаев спонтанная термическая полимеризация обусловлена, инициированием перекисями, которые легко образуются на свету даже при кратковременном контакте мономеров с кислородом воздуха.
20) Кинетика поликонденсации
Рассмотрим основные кинетические закономерности поликонденсации на примере полиэтерификации. Катализаторами реакции этерификации являются кислоты и щелочи. Механизм:
1. Протонирование кислоты-реагента кислотой-катализатором (НА):
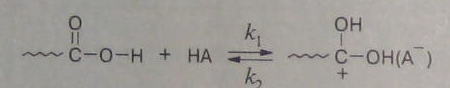
2. Атаки протонированным реагентом гидроксильной группы спирта с последующим распадом интермедиата до продуктов реакции:

Если в данной реакции удалять воду, то можно учитывать лишь прямое направление реакции. Тогда:
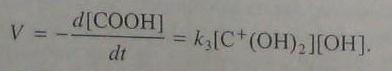 (5.96)
(5.96)
Неопределяемая величина [С+(0Н)2] может быть исключена с помощью выражения (5.97):
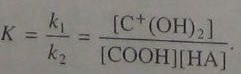
В результате получаем:
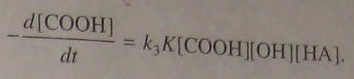 (5.98)
(5.98)
В отсутствии внешнего катализатора его функции кислота-мономер. Тогда:
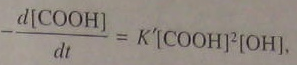 (5.99)
(5.99)
Где К’=к3К. тк при поликонденсации концентрации разных функциональных групп обычно равны (в целях получения ВМС полимера), то уравнение (5.99) упрощается:
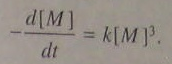 (5.100) интегрируем:
(5.100) интегрируем: 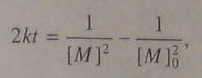 (5.101)
(5.101)
Используем понятие степени завершенности реакции: 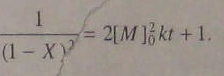 (5.102)
(5.102)
Уравнение (5.102), исходящее из третьего порядка скорости по концентрации мономера, достаточно хорошо описывает экспериментальные данные. Встречающиеся отклоняя при средних и глубоких степенях завершенности связаны с изменением состояния реакционной среды - уменьшением полярности, вследствие исчерпания карбоксильных и гидроксильных (или других полярных) групп мономеров, и возрастанием вязкости.
Степень ступенчатой полимеризации равна числу мономерных звеньев в цепи. При ступенчатой полимеризации двух нефункциональных мономеров A-R-A и B-R-B степень полимеризации равна половине их количества в цепи. Сочетание (5.92) и (5.102) приводит к зависимости степени полимеризации от времени:

Из этого уравнения следует, что темп нарастания молекулярной массы продукта поликонденсации уменьшается со временем.
Изложенное выше касалось самокатализируемой поликонденсации. При наличии внешнего катализатора, уравнение скорости отвечает второму порядку по концентрации мономера:
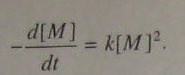 (5.104)
(5.104)
Интегрируем и получаем следующие уравнения с использование (5.92) и (5.106)
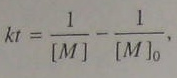 (5.105)
(5.105) 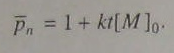 (5.106)
(5.106)
Из уравнения (5.106) следует, что при катализируемой поликонденсации имеет место линейная зависимость степени полимеризации от времени.
На практике самокатализируемая реакция используется при получении полиамидов, тогда как при получении полиэфиров и фенопластов - продуктов поликонденсации фенола с формальдегидом - применяется катализатор. При предыдущем изложении предполагалось равенство концентраций мономеров, что является одним из условий получения высокомолекулярного полимера при поликонденсации. Количественной мерой, отражающей степень эквивалентности концентраций мономеров, является параметр
 (5.107)
(5.107)
вследствие [М1]>[М2]. Связь среднечисловой степени полимеризации с параметром эквивалентности дается выражением: 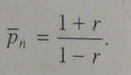 (5.108)
(5.108)
При строго эквивалентных количествах функциональных групп на концах макромолекул реакция между ними может продолжаться сколь угодно долго, теоретически - вплоть до образования гигантской макромолекулы. Из этого следует, что для стабилизации молекулярной массы полимера небольшой избыток одного из мономеров может оказаться полезным. В этом случае все макромолекулы будут иметь одинаковые функциональные группы, например А--А, что исключает возможность реакции между ними. Для этих целей используют также малые добавки монофункционального соединения. При этом уравнение (5.108) по-прежнему применимо, однако параметр r рассчитывается по-другому. При поликонденсации двух гомофункциональных мономеров ARA и BR1B в присутствии монофункционального соединения R2B
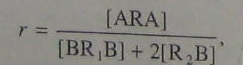 (5.109)
(5.109)
где [ARA] = [BR1B]. Коэффициент 2 вводится потому, что эффект добавки R2B аналогичен эффекту избытка бифункционального мономера BR1B, a уравнение (5.109) получено применительно к реакции двух бифункциональных мономеров. При поликонденсации одного гетерофункционального мономера ARB в присутствии BR1
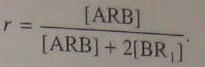
21)Особенности синтеза полимеров методом поликонденсации.
Поликонденсация - процесс образования полимеров из би- или полифункциональных мономеров, который чаще всего сопровождается выделением побочных низкомолекулярных соединений (воды, спирта и т.д.). Является одним из видов ступенчатого синтеза полимеров.
Различают линейную и т рехмерную поликонденсацию.
Поликонденсация, в которой участвуют только бифункциональные мономеры, приводит к образованию линейных молекул полимера и называется линейной.
Процесс поликонденсации, в котором участвуют молекулы с тремя или большим числом функциональных групп, приводит к образованию разветвленных или трехмерных (сетчатых, сшитых) структур и называется трехмерной поликонденсацией.
Если обозначить через f функциональность, т. е. число функциональных групп в молекуле реагирующего вещества, а через N0 число исходных молекул, то общее число функциональных групп в молекулах реагирующих веществ будет равно Nof.
Обозначив через N число молекул конечного продукта реакции, получим следующее выражение для степени завершенности реакции:
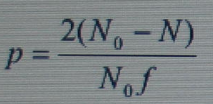
Величина 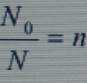 соответствует среднему коэффициенту поликонденсации, откуда
соответствует среднему коэффициенту поликонденсации, откуда
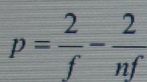
Если п очень велико, то p = 2/ f. Тогда для бифункциональных соединений (f =2)
степень завершенности реакции р = 1.
Для трифункциональных соединений p =2/3, для тетрафункциональных p = ½ и т. д.
Следовательно, во всех случаях, когда р<1, происходит образование пространственной структуры.
1. Линейная поликонденсация
На свойства получаемых полимеров большое влияние оказывает функциональность исходных соединений. Если оба мономера бифункциональны, то при поликонденсации образуются строго линейные высокомолекулярные соединения, вещества, растворимые в растворителях. 




Во второй стадии уже начинается образование сетки, полимер нерастворимый, но мягкий и эластичный, в третьей стадии -- неплавкий и нерастворимый. Вторую, переходную стадию не всегда можно заметить. Иногда одни и те же исходные вещества в зависимости от соотношения образуют полимеры различной структуры.
 В процессе трёхмерной поликонденсации происходит образование трёхмерной структуры – нерастворимого геля. Реакционная смесь разделяется на две части: нерастворимый гель и растворимый золь, который может быть отделён от геля путём экстракции растворителя. Этот момент называют точкой гелеобразования. Нерастворимость геля объясняется тем, что он представляет собой единю пространственную сетку, в которой отдельные цепи связаны между собой химически настолько прочно, что растворитель не в состоянии отделить их друг от друга. Такое отделение было бы равноценно деструкции полимера. Растворение возможно только в том случае, когда активность растворителя достаточна, чтобы вызвать химическое расщепление отдельных связей и химическое изменение природы полимера.
В процессе трёхмерной поликонденсации происходит образование трёхмерной структуры – нерастворимого геля. Реакционная смесь разделяется на две части: нерастворимый гель и растворимый золь, который может быть отделён от геля путём экстракции растворителя. Этот момент называют точкой гелеобразования. Нерастворимость геля объясняется тем, что он представляет собой единю пространственную сетку, в которой отдельные цепи связаны между собой химически настолько прочно, что растворитель не в состоянии отделить их друг от друга. Такое отделение было бы равноценно деструкции полимера. Растворение возможно только в том случае, когда активность растворителя достаточна, чтобы вызвать химическое расщепление отдельных связей и химическое изменение природы полимера.
В точке гелеобразования среднечисловая молекулярная масса невелика, среднемассовая молекулярная масса стремится к бесконечности.
После достижения точки гелеобразования количество золя начинает быстро убывать вследствие его перехода в гель. Вязкая реакционная масса сначала превращается в эластичный материал, а потом в твёрдый неплавкий и неастворимый продукт. Наряду с межмолекулярными процессами может идти реакция между функциональными группами одной сетчатой структуры.
Способы проведения поликонденсации
1 Поликонденсация в расплаве.
Процесс проводят при температурах порядка 200-280˚С в токе инертного газа. Заканчивают его в вакууме с целью более полного удаления низкомолекулярных веществ (аммиак, вода, хлороводород). Положительные стороны поликонденсации в расплаве:
высокий выход полимера; высокая степень чистоты полимера; возможность получения полимеров с пониженной реакционной способностью; простота технологической схемы;
возможность использовать полимер в виде расплава для формования волокон и пленок.
Однако процесс имеет достаточно большую продолжительность. Необходимость проведения его при высоких температурах может вызывать деструкцию образующегося полимера. Для такого способа поликонденсации нужно использовать термически стойкие мономеры.
Например, синтез полиэфиров из гликолей и дикарбоновых кислот, а также из оксикислот проводят, главным образом, в расплаве исходных веществ при сравнительно высоких температурах (170—250°С) в токе инертного газа при обычном давлении и заканчивают в вакууме. Избыток одного из компонентов приводит к уменьшению средней молекулярной массы полимера вследствие блокирования концевых групп полимера и протекания реакций ацидолиза или алкоголиза.
2 Поликонденсация в растворе.
Мономеры при таком способе проведения поликонденсации находятся в растворенном состоянии. При поликонденсации в растворе существенное влияние на скорость реакции и молекулярную массу оказывают концентрация реагирующих веществ и природа растворителя. Добавляя один из мономеров к раствору второго или смешивая отдельные растворы их в одинаковых или различных растворителях, можно в зависимости от дозировки регулировать молекулярную массу полимера.
Иногда применяют растворители, не смешивающиеся с исходными мономерами, при условии образования в дальнейшем гомогенной системы. В этих случаях выпавшие в осадок полимеры, сохранив свои концевые функциональные группы, длительное время не теряют активности и могут поэтому быть использованы для синтеза блок-сополимеров.
Этот метод позволяет проводить поликонденсацию в жидкой фазе и добиться высоких молекулярных масс без нагревания до температур, вызывающих деструкцию полимеров.
Положительные стороны поликонденсации в растворе:возможность осуществлять процесс в мягких условиях; хорошая теплопередача - отсутствие местных перегревов;
низкая вязкость - можно легко удалять низкомолекулярные побочные продукты, например, в виде азеотропа;
Минусами процесса являются: необходимость тщательной очистки растворителя перед синтезом; определенные трудности выделения самого полимера и остатков растворителя из него.
Поликонденсация в растворе представляет определенный интерес для тех производств, где исходный полимер находится в растворе (лаки, краски, прядильные растворы, пленки).
3 Межфазная поликонденсация
Разработан метод поликонденсации на поверхности раздела двух несмешивающихся жидкостей - так называемый метод межфазной поликонденсации. Это гетерогенный необратимый процесс. Скорость его лимитируется скоростью диффузии реагентов к границе раздела фаз. Перед проведением поликонденсации исходные реагенты растворяют раздельно. В этом процессе в случае взаимодействия двухатомного фенола с дихлорангидридом дикарбоновой кислоты дихлорангидрид растворяют, например, в бензоле, а двухатомный фенол - в воде. При контакте приготовленных растворов на границе раздела фаз мгновенно образуется полимер. Полимер удаляют из зоны реакции. Процесс ведут до полного исчерпания мономеров. Для увеличения поверхности контакта компоненты фаз перемешивают.
При интенсивном перемешивании двух жидкостей (скорость вращения мешалки около 4000 об/мин) поликонденсация протекает со скоростью ионных реакций даже при комнатной температуре. Для связывания выделяющегося хлористого водорода целесообразно вводить в реакционную смесь щелочные добавки (NaOH или Ме2СОз). Благодаря высокой скорости реакции соотношение исходных компонентов перестает играть существенную роль. Поскольку реакция протекает на поверхности раздела двух несмешивающихся жидкостей, молекулярная масса образующегося полимера зависит от величины этой поверхности и скорости перемешивания, а также характера растворителя, концентрации растворов, рН среды и строения исходных мономеров. Применение инертного газа необязательно.
По описанному методу исходные мономеры необходимо применять в виде растворов очень низких концентраций, что затрудняет получение больших количеств полимера при межфазной поликонденсации. Наиболее целесообразно применять мономеры с высокой реакционной способностью (дихлорангидриды дикарбоновых кислот, диамины, дифенолы). В этом случае время контакта резко уменьшается. Низкая температура, обычно комнатная, дает возможность уменьшить побочные реакции. При увеличении температуры молекулярная масса полимера и выход уменьшаются. Полимеры, полученные на границе раздела двух фаз, как правило, имеют более высокую молекулярную массу, чем полимеры, получаемые в расплаве и особенно в растворе. Так, молекулярная масса полиэфиров, синтезированных в расплаве, не превышает 20 000, а при синтезе полиэфиров методом межфазной поликонденсации молекулярная масса их может достигать 1 000 000. Большая молекулярная масса получаемых полимеров, вероятно, обусловлена тем, что макромолекулы, пока они сравнительно невелики, остаются в растворе и продолжают расти. Только после достижения достаточно больших размеров они переходят в осадок.
Простота, высокие скорости межфазной поликонденсации позволяют легко осуществлять её непрерывными методами. Положительными сторонами процесса также являются низкая температура проведения процесса, не обязательная высокая степень очистки реагентов.
Минусами процесса являются: использование дорогостоящих мономеров с высокой реакционной способностью, а также большие затраты на очистку (регенерацию) растворителей.
Метод применяется тогда, когда другие способы невозможны. Например, таким способом можно получать высокодисперсные полимерные порошки из термически нестойких мономеров.
22. Полимераналогичные превращения целлюлозы.
ПОЛИМЕРАНАЛОГИЧНЫЕ ПРЕВРАЩЕНИЯ - химические р-ции функциональных групп макромолекул или отдельных атомов основной цепи, в ходе которых длина и строение скелета цепи сохраняются, но изменяются состав и строение боковых групп. Реакционная способность функциональных групп макромолекул в полимераналогичных превращениях, в соответствии с допущением Флори, не зависит от длины цепи, с которой эта группа связана, и во многих случаях не отличается от реакционной способности соответствующих низкомолекулярных аналогов. Целлюлоза является основной составной частью природных и искусственных целлюлозных волокон.
Химические превращения целлюлозы
Путем полимераналогичных превращений из целлюлозы получают три основных класса ценных полимерных материалов:
Сложные эфиры целлюлозы - ацетаты - получаются при действии на целлюлозу уксусного ангидрида в присутствии катализатора - серной или хлорной кислоты или их смесей:

Нитраты целлюлозы. Нитраты получают действием на целлюлозу смеси азотной и серной кислот:
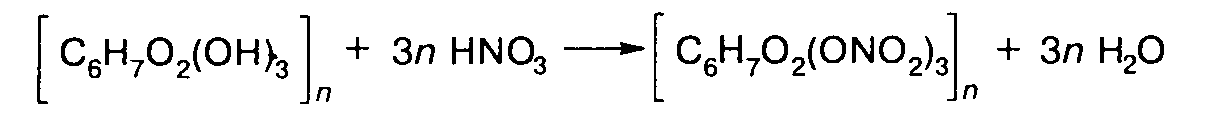
Реакция протекает, начиная с 0 °С. Степень замещения определяется только составом нитрующей смеси.
Простые эфиры целлюлозы. При их получении целлюлоза предварительно активируется путем обработки щелочью, при этом она набухает. В качестве алкилирующих агентов используются алкилгалогениды, алкилсульфа-ты и др. Реакция образования метилцеллюлозы протекает при 80-100 °С и под давлением:
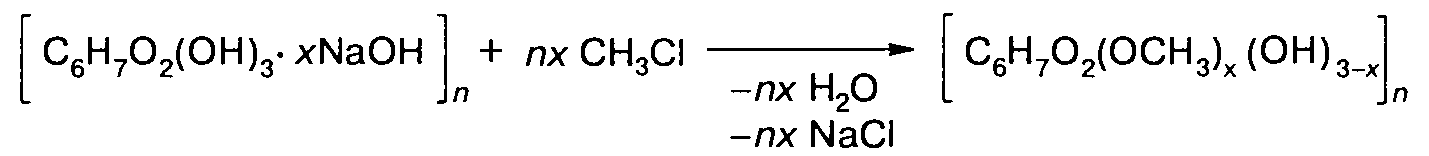
Выход реакции увеличивается с увеличением давления и с уменьшением температуры.
Карбоксиметилцеллнэлоза. Карбоксиметилцеллюлоза получается при взаимодействии щелочной целлюлозы с монохлоруксусной кислотой или ее натриевой солью:
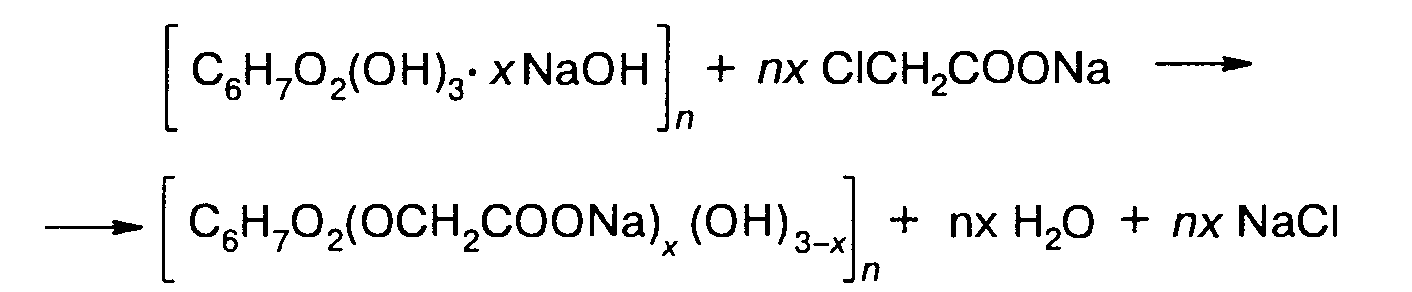
Экзотермическая реакция протекает за 1,5-2 ч при самопроизвольном повышении температуры от 20 до 40 °С.
При обработке щелочной целлюлозы окисью этилена в мягких условиях при 33-40 °С получается оксиэтилцеллюлоза.
Структурная модификация целлюлозы
Гидратцеллюлоза аналогична по составу исходной целлюлозе, отличается от нее расположением звеньев и большей степенью гидратации полярных групп. Гидратцеллюлоза получается двумя методами: физическим и химическим. В первом случае целлюлозу растворяют и снова осаждают. Во втором путем полимераналогичной реакции целлюлозу переводят в одно из ее производных, затем последнее в результате реакции гидролиза вновь переводят в целлюлозу. Оба метода приводят к структурной модификации целлюлозы. Последним методом производится вискозное или медноаммиачное волокно - первое искусственное волокно, полученное человеком. Технология, разработанная в 20-30-х годах, включает две стадии. На первой - целлюлоза последовательно обрабатывается раствором щелочи и сероуглеродом:

Образовавшийся ксантогенат растворяют в разбавленном растворе щелочи и затем в кислой среде проводят обратную реакцию, приводящую к образованию гидратцеллюлозы:
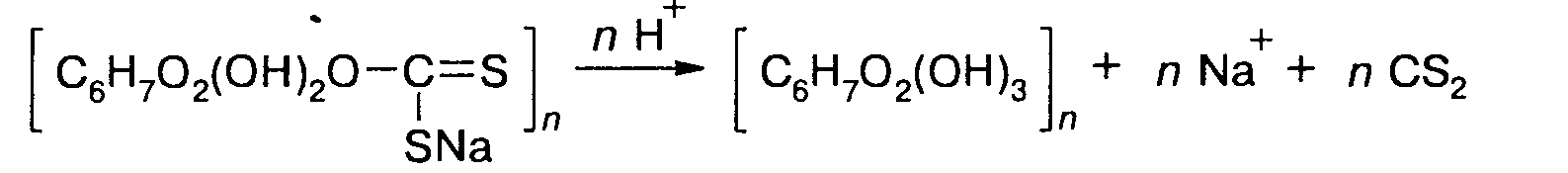
23. Реакционная способность мономеров и радикалов в радикальной полимеризации. Гель-эффект.
Интернет: Реакционная способность мономеров в радикальной сополимеризации определяется сопряжением, полярностью, степенью экранирования двойной связи заместителями. С увеличением энергии сопряжения в мономере возрастает его активность в реакциях присоединения к радикалам вследствие резонансной стабилизации переходного состояния. По этой же причине увеличивается стабильность образующихся радикалов. Следовательно, ряды активностей мономеров противоположны рядам активностей соответствующих радикалов, что составляет существо правила антибатности. При этом сопряжение с заместителем снижает активность радикала в значительно большей степени, чем повышает активность мономера. Поэтому мономер, более активный в сополимеризазии, в гомополимеризации кажется менее активным (полимеризуется с меньшей скоростью).
Семчиков: Параллельно с развитием количественной теории сополимеризации полвека назад была разработана количественная схема Алфрея-Прайса, связывающая константы сополимеризации с эмпирическими параметрами реакционной способности. Согласно этой схеме, константа скорости роста в радикальной полимеризации и сополимеризации выражается эмпирическим уравнением:

где Pi и Qj - параметры, учитывающие резонансный; еi и еj - полярный факторы реакционной способности. Исходя из (6.96), легко могут быть получены выражения для относительных активностей мономеров:

Далее, перемножив относительные активности мономеров в (6.97) и логарифмируя полученное произведение, получаем:

откуда следует, что тенденция к чередованию при сополимеризации определяется лишь разницей значений полярных параметров мономеров.
Схема Q-e широко используется в сополимеризации, так как она позволяет рассчитать относительные активности мономеров и, следовательно, состав и структуру сополимера, не проводя сополимеризации, по известным значениям Q и е мономеров. Значение схемы Q-e состоит также в том, что она позволила отнести мономеры к определенным группам, исходя из значений параметров Q и е: активным (Q > 0,5) и неактивным (Q < 0,1), электронодонорным (е < 0) и электроноакцепторным (е>0) и, тем самым, предсказать тип полимеризационного процесса, в котором целесообразно использовать данный мономер.
Анализ систематических данных в области радикальной (со)полимеризации приводит к выводу о том, что реакционная способность мономеров и радикалов в реакции роста определяется резонансной стабилизацией сопряжением, полярностью двойной связи, а также степенью ее экранирования заместителями.
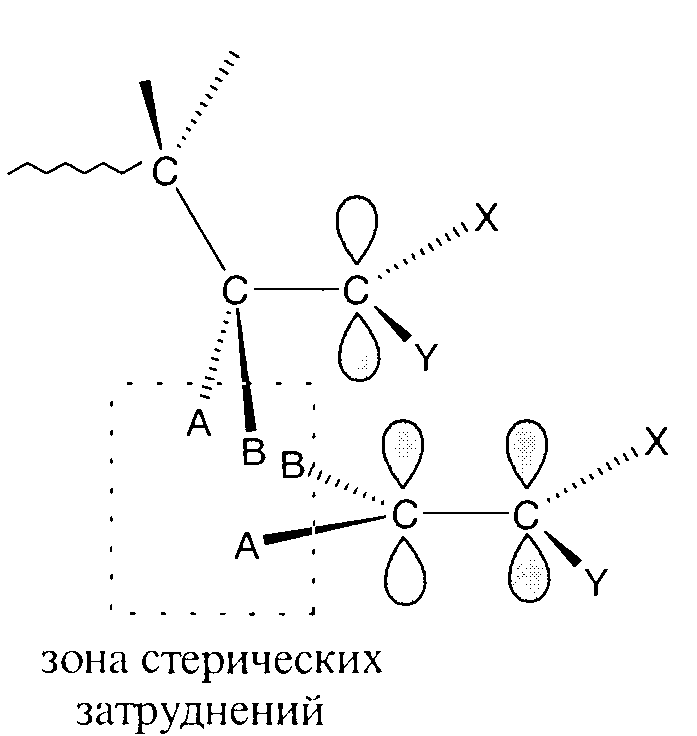 Стерический фактор. Значение стерического фактора проявляется особенно ярко в реакциях радикального присоединения дизамещенных этилена. Пространственная структура органических соединений в значительной степени определяется типом гибридизации атомов углерода. Ненасыщенные атомы радикала роста и мономера имеют sp2-гибридизацию. Это означает, что оси p-орбиталей ненасыщенных атомов перпендикулярны плоскости, в которой расположены сигма - связи. Атомы углерода основной цепи радикала образуют плоский зигзаг, все они, за исключением концевого ненасыщенного атома углерода, имеют sp3-гибридизацию. Из приведенной ниже схемы видно, что при сближении условного тетразамещенного мономера (АВ)С = C(XY) со «своим» радикалом роста вероятно контактное взаимодействие, т. е. отталкивание заместителей А и В мономера и атома углерода радикала до совмещения осей p-орбиталей. В результате реакция роста не может осуществиться.
Стерический фактор. Значение стерического фактора проявляется особенно ярко в реакциях радикального присоединения дизамещенных этилена. Пространственная структура органических соединений в значительной степени определяется типом гибридизации атомов углерода. Ненасыщенные атомы радикала роста и мономера имеют sp2-гибридизацию. Это означает, что оси p-орбиталей ненасыщенных атомов перпендикулярны плоскости, в которой расположены сигма - связи. Атомы углерода основной цепи радикала образуют плоский зигзаг, все они, за исключением концевого ненасыщенного атома углерода, имеют sp3-гибридизацию. Из приведенной ниже схемы видно, что при сближении условного тетразамещенного мономера (АВ)С = C(XY) со «своим» радикалом роста вероятно контактное взаимодействие, т. е. отталкивание заместителей А и В мономера и атома углерода радикала до совмещения осей p-орбиталей. В результате реакция роста не может осуществиться.
Резонансный фактор. В зависимости от наличия или отсутствия сопряжения двойной связи мономера с ненасыщенной группой заместителя все мономеры делятся на активные и неактивные. Типичные представители каждой группы представлены ниже:
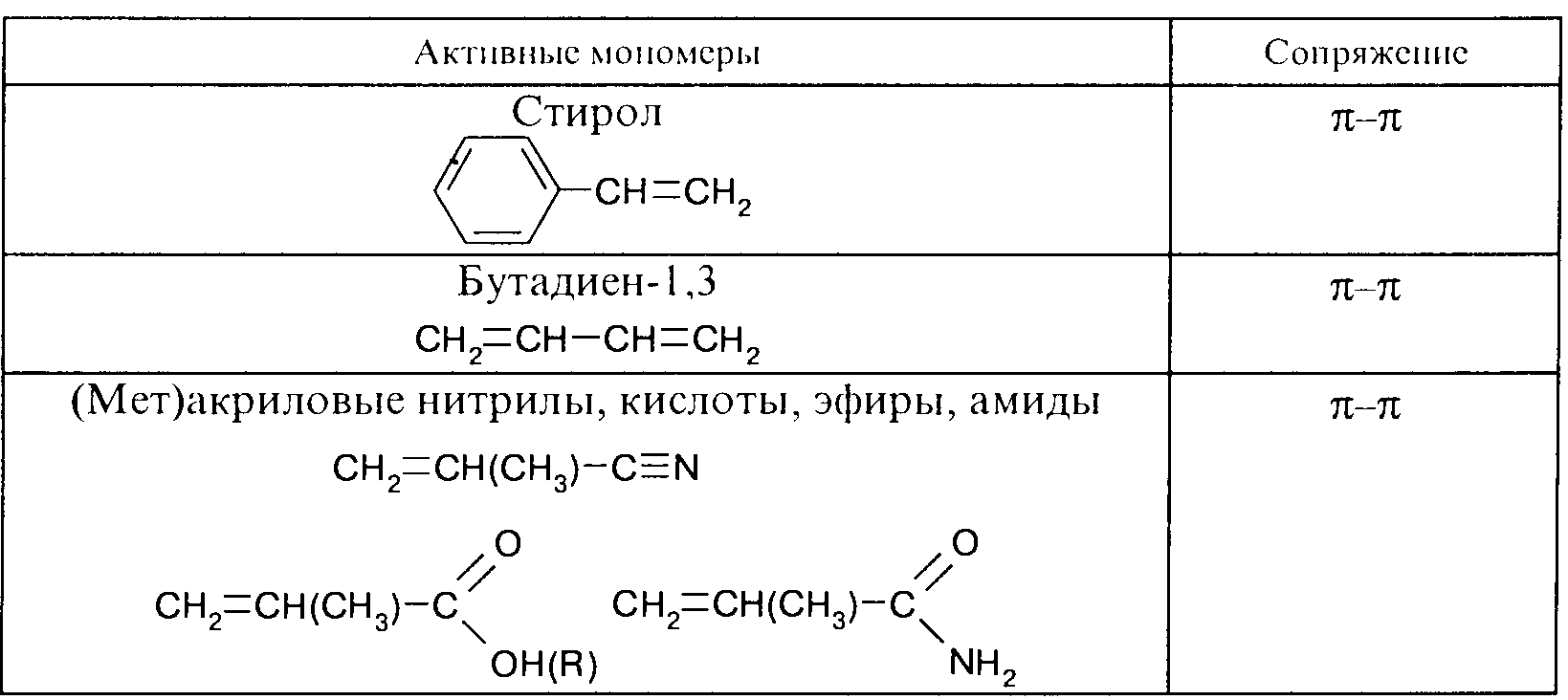

Сопряжение или резонансный эффект противоположным образом влияет на реакционную способность мономеров и радикалов - увеличивает активность первых и уменьшает активность вторых. Из этого следует, что ряды реакционной способности мономеров и соответствующих им радикалов противоположны. Это положение известно как правило антибатности.
Влияние сопряжения на реакционную способность мономеров и соответствующих им радикалов роста неодинакова по эффективности - активность мономера возрастает в меньшей степени по сравнению со стабилизацией, т. е. уменьшением реакционной способности радикала.
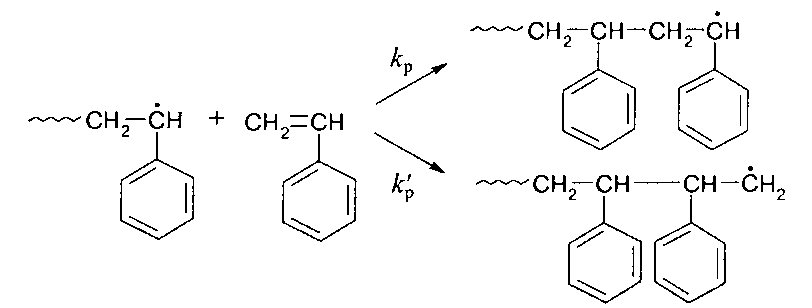 Схема, на которой приведены два возможных направления реакции роста при сополимеризации стирола:
Схема, на которой приведены два возможных направления реакции роста при сополимеризации стирола:
В первом случае реализуется сопряжение ароматического заместителя с образующимся радикалом и переходным комплексом, и поэтому мономер ведет себя как активный. Во втором случае сопряжение отсутствует, поскольку ненасыщенный атом углерода радикала разделен с ароматическим заместителем двумя сигма-связями, и в этом случае мономер является неактивным. В результате первая реакция оказывается более предпочтительной (кр » к'р) и присоединение радикала к мономеру происходит с вероятностью, большей 90 % по типу «голова» к «хвосту».
Атом углерода мономера, атакуемый радикалом, изменяет гибридизацию sp2 на sp3 и, таким образом, выбывает из системы сопряжения. Энергия, необходимая для этого, и называется энергией локализации мономера Lß. Аналогичное рассуждение можно провести и по отношению к сопряженному радикалу, однако энергия локализации радикала La не оказывает существенного влияния на относительные активности мономеров.
С увеличением параметра Q абсолютное значение энергии локализации мономера падает. Это означает, что с увеличением энергии сопряжения в мономере снижается энергия, необходимая для активации разрыва его двойной связи.
Реакционная способность мономеров и радикалов, определяемая лишь резонансным фактором, называется идеальной реакционной способностью.
Полярный фактор. Двойная связь мономеров, подверженных радикальной сополимеризации, как правило, является поляризованной вследствие донорно-акцепторного действия заместителей, как и ненасыщенный атом углерода радикала роста:

Донорно-акцепторный эффект заместителей приводит к возникновению частичных зарядов на ß-атоме углерода двойной связи и а-атоме углерода концевого звена радикала роста (ненасыщенном).
Влияние полярного фактора реакционной способности наиболее ярко проявляется в радикальной сополимеризации, где он ответственен за возникновение эффекта чередования мономерных звеньев. Впервые на значение полярного фактора в сополимеризации обратил внимание Прайс, который сделал заключение о том, что «легче всего сополимеризация протекает в таких бинарных системах, в которых один мономер имеет избыток, другой -недостаток электронов».
Гипотеза, получившая широкое распространение к настоящему времени, объясняла склонность к чередованию звеньев при сополимеризации переносом электрона между компонентами переходного комплекса, т.е. является развитием гипотезы Уол-линга:
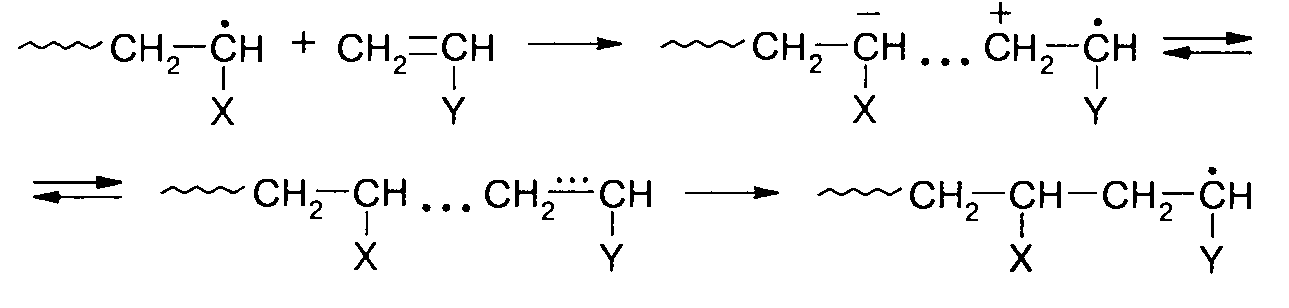
В приведенной схеме мономер СНг=СНХ, например метилметакрилат, и соответствующий ему радикал роста являются акцепторами электрона, а мономер СНг=СНУ, например стирол, является донором электрона. Считается, что вклад ионной структуры в переходное состояние снижает энергию активации перекрестного роста, в результате при сополимеризации проявляется тенденция к чередованию мономерных звеньев, однако, образующийся сополимер остается статистическим. Гель-эффект.
Возросшим периодом существования макрорадикалов объясняется явление резкого самоускорения полимеризации на более поздних этапах ее — гель-эффект, или эффект Тромсдорфа*. Гель-эффект связан с диффузионными затруднениями, обусловленными быстрым увеличением вязкости среды и, как правило, сопровождается повышением молекулярной массы полимера. Величина наблюдаемого самоускорения и глубина реакции, при которой оно наступает, колеблется в довольно широких пределах и зависит от природы мономера, молекулярной массы полимера, температуры полимеризации, наличия инертных растворителей и т. д. У метилакрилата при 30°С самоускорение заметно уже при глубине полимеризации, меньшей 1%, у метилметакрилата (30°С) — при 15%, а у стирола (50°С) — при 50%. По мере течения полимеризации одновременно возрастает скорость ее, которая у метилметакрилата (50°С) достигает максимума при глубине реакции порядка 60%. Сразу же после этого вся реакционная масса застывает в гель и скорость полимеризации резко падает, так как передвижение мономерных молекул в такой вязкой системе встречает большое сопротивление; при снижении содержания мономера в геле приблизительно до 15% реакция прекращается. Однако она может быть практически доведена до конца, если разбавить систему инертным растворителем.
Особенно велико значение гель-эффекта в тех случаях, когда гелеобразование наступает на ранних стадиях полимеризации, например при получении сетчатых полимеров.
Гель-эффект во многом напоминает гетерофазную полимеризацию, при которой процесс образования полимера протекает в двух или более фазах (или на поверхности, разделяющей их). У гетерофазной полимеризации, как и при гель-эффекте, наблюдается в ходе реакции самоускорение и возрастание молекулярной массы, обусловленные накоплением в системе долгоживущих радикалов.
25. Классификация и гидродинамические свойства полиэлектролитов.
Полиэлектролитами называют высокомолекулярные соединения, макромолекулы которых содержат ионогенные группы, способные к диссоциации на ионы. В зависимости от природы и степени диссоциации ионогенных групп полиэлектролиты делятся на сильные и слабые поликислоты и полиоснования, а также на полиамфолиты. Полиэлектролиты последнего типа содержат основные и кислотные группы. Известно много синтетических и природных полиэлектролитов. Из последних особенно большое значение имеет белок, который образуется из аминокислот различного строения и содержит полипептидную цепь с повторяющейся группировкой -CO-NH-. Некоторые из заместителей полипептидной цепи содержат кислотные и основные группы, поэтому белок является полиамфолитом. Ниже приведены примеры отдельных полиэлектролитов:
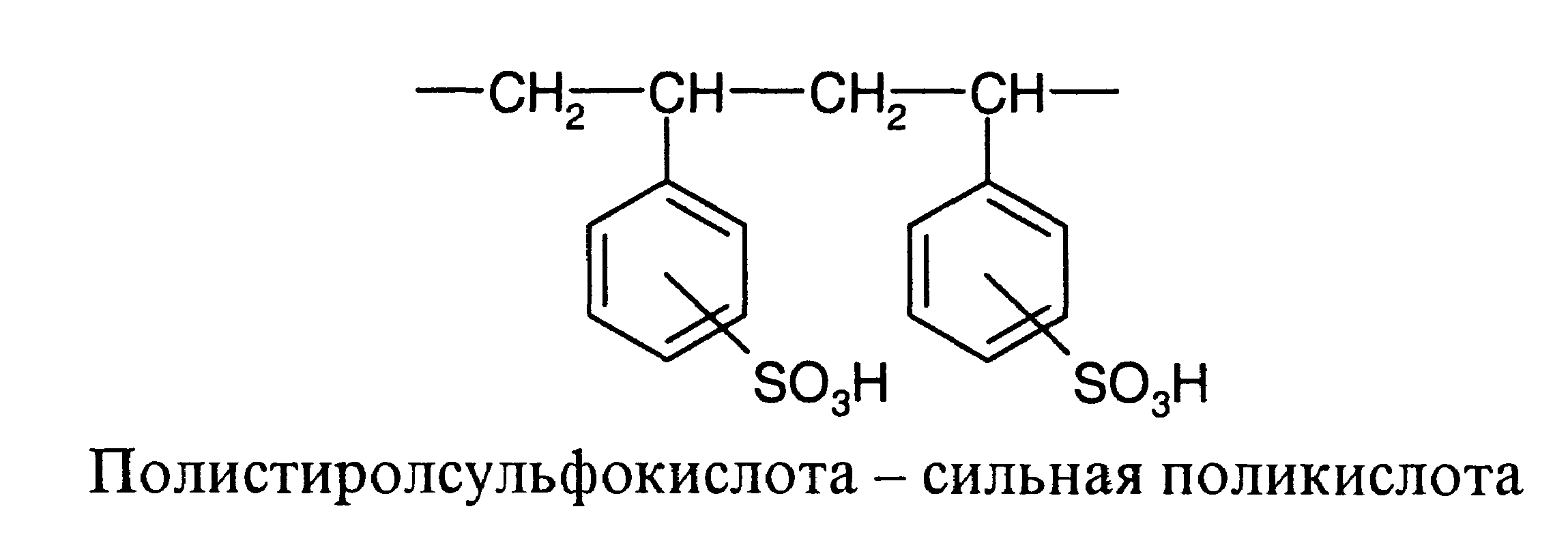
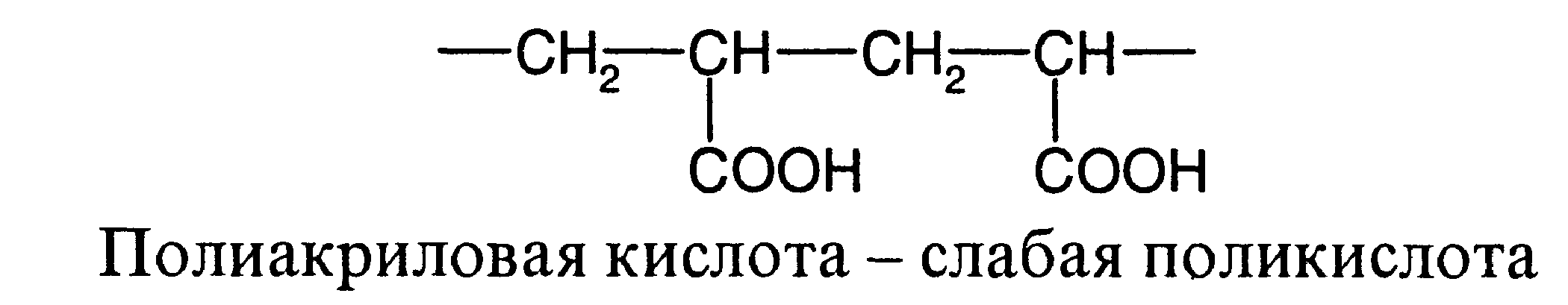
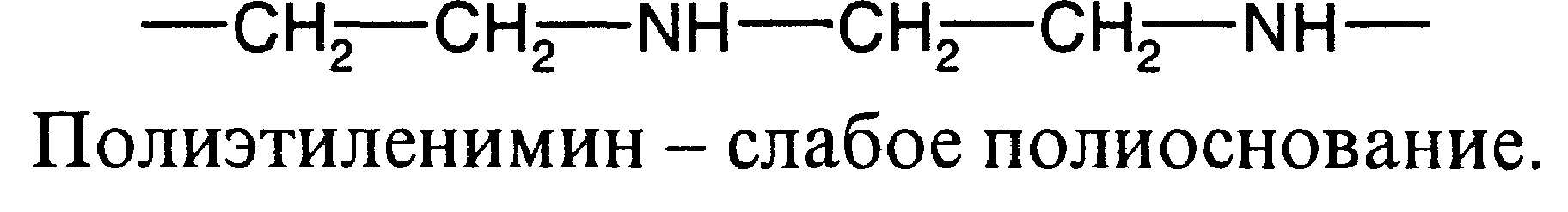

Свойства заряженных макромолекул качественно отличаются в зависимости от суммарной величины заряда каждой макромолекулы: различают сильно заряженные полиэлектролиты, например ДНК, и слабо заряженные (слабые полиэлектролиты), например полиакриловая кислота. В сильно заряженных макромолекулах ионизирована большая часть звеньев, поэтому их свойства в основном определяются электростатическими (кулоновскими) взаимодействиями. В слабо заряженных макромолекулах ионизирована меньшая часть звеньев, их свойства определяются как электростатическими, так и неэлектростатическими взаимодействиями. Для водных растворов так называемое гидрофобное взаимодействие, играющее большую роль в биологических системах. Под этим термином подразумеваются силы притяжения между неполярными группами в полярной водной среде. Чередование микрообластей с полярными и неполярными взаимодействиями приводит к появлению регулярных неоднородностей в растворах полиэлектролитов.
Свойства растворов полиэлектролитов
Наиболее ярко влияние зарядов проявляется при изучении вязкостных свойств полиэлектролитов. Рассмотрим зависимость вязкости раствора желатины (белка) от рН среды. Минимальной вязкостью обладают растворы при рН 4,7, отвечающем изоэлектрической точке. В изоэлектрическом состоянии число диссоциированных кислотных и основных групп макромолекулы амфолита равно и минимально, вследствие чего суммарный заряд макромолекулы равен нулю. При этих условиях для цепи характерны свернутые конформации. При изменении рН (по сравнению с рН изоэлектрической точки), вызванном добавкой низкомолекулярного электролита (кислоты или щелочи), степень диссоциации ионогенных групп желатины увеличивается. Увеличение рН приводит к диссоциации кислотных групп (а), уменьшение рН вызывает диссоциацию основных групп (б):

В обоих случаях цепь разворачивается в результате возникновения и отталкивания одноименных зарядов в ней. Это приводит к значительному возрастанию вязкости раствора. По достижении определенной степени диссоциации кислотных или основ