Лекция №12
БОЛЕЗНИ СИСТЕМЫКРОВИ
Анемия (малокровие) – это патологическое состояние организма, при котором уменьшается число эритроцитов в единице объема крови.
Классификация анемий: (Мосягина, 1979г.)
1. Дефицитные анемии:
1. железодефицитные
2. витаминодефицитные
3. протеинодефицитные
2. Гипо- и апластические анемии
1. врожденные
2. приобретенные
3. Геморрагические анемии
1. вследствие острой кровопотери
2. вследствие хронической кровопотери
4. Гемолитические анемии
1. врожденные
2. приобретенные
5. анемии при различных заболеваниях (вторичные, сопутствующие).
АНЕМИЯ ЖЕЛЕЗОДЕФИЦИТНАЯ. Этиология: Наиболее частой причиной является алиментарный дефицит железа, большая кровопотеря (кровотечения), инфекционные болезни.
Клиническая картина. Характерны снижение аппетита, замедленная прибавка массы тела, бледность кожных покровов, мышечная гипотония.
При аускультации сердца выявляют систолический (анемический) шум. При глубокой анемии поражаются эпителиальная ткань (шершавая кожа, ложкоподобные хрупкие ломкие ногти, выпадение волос) и слизистые оболочки (атрофия сосочков языка, эрозии в углах рта). Количество гемоглобина значительно меньше нормы, в то время как число эритроцитов снижается умеренно.
Прогноз. При своевременном активном лечении благоприятный.
Лечение. Рациональное питание с достаточным содержанием белков, витаминов, железа. Препараты железа назначают внутрь в сочетании с аскорбиновой кислотой. Хороший эффект оказывают сульфатные формы железа — таблетки «Феррокаль», «Ферроплекс», конферон, а также «Гемостимулин» — препарат, содержащий молочнокислое железо (50 %), сернокислую медь (1 %), глюкозу (20 %) и сухой гематоген (25 %). Препараты железа целесообразно принимать в промежутках между приемами пищи.До и после приема железа не рекомендуется давать детям чай, жирные и некоторые мучные продукты во избежание образования нерастворимых соединений, ухудшающих усвоение железа. Курс лечения не менее 1,5— 2 мес. При непереносимости пероральных препаратов и расстройствах всасывания может быть применен ферковен (смесь трехвалентного железа и глюконата кобальта в растворе углеводов), феррум Лек. Лечение ферковеном (внутривенные инъекции по 1—2 мл через день) проводится только в стационаре, поскольку возможны отрицательные реакции.
АНЕМИЯ витаминодефицитная. Этиология и патогенез. Нарушение созревания эритроцитов, вызванное дефицитом витамина В12, фолиевой кислоты, которое может развиться при воздействии: частых инъекций, неполноценного питания, вскармливания козьим молоком (у детей младшего возраста), целиакии (нарушение всасывания из кишечника), хронических болезней желудочно-кишечного тракта и печени, глистной инвазии
Клиническая картина. Ухудшается аппетит, дети не прибавляют в массе. Кожа и слизистые оболочки бледные, с субиктеричным оттенком, иногда геморрагии на коже. Течение хроническое.
Диагноз. Устанавливают на основании клинической картины, картины периферической крови и костного мозга.
Прогноз. Зависит от основного заболевания. В ряде случаев благоприятный.
Лечение. Показаны витамины В и фолиевая кислота в возрастной дозировке; большие дозы аскорбиновой кислоты, препараты печени; диета, богатая белками, с ограничением жиров; лечение основного заболевания; при воспалительных процессах — антибиотики.
АНЕМИЯ ГИПОПЛАСТИЧЕСКАЯ.
Этиология и патогенез. Принято различать две основные группы гипопластических анемий: врожденные (конституциональная анемия Фанкони). Приобретенные в результате воздействия экзогенных факторов (физические (лучевая энергия), химические (красители, бензены), лекарственные (хлорэтиламины, антиметаболиты, сульфонамиды, некоторые антибиотики).
Клиническая картина. К ранним симптомам относятся общая слабость, утомляемость, боли в костях и суставах, геморрагический синдром (носовые кровотечения, кровоизлияния в кожу). Постепенно нарастают бледность кожи и слизистых оболочек. При врожденных формах гипо- и апластической анемии кожа имеет характерный пепельный оттенок. Печень несколько увеличена.
Прогноз. В большинстве случаев неблагоприятный. Зависит от степени поражения костного мозга и фазы патологического процесса. Положительный эффект может быть получен при раннем проведении спленэктомии у больных с подострыми и хроническими формами заболевания.
Лечение. Патогенетической терапии не существует. В остром периоде применяют глюкортикостероидные гормоны в больших дозах; трансфузии эритроцитной массы 1—2 раза в неделю с заместительной целью. Ранняя спленэктомия дает благоприятный результат. Показано длительное, но прерывистое применение анаболических препаратов (ретаболил, метиландростенолон). Наибольшие надежды в лечении тяжелой апластической анемии связаны с пересадкой костного мозга от строго подобранного по совместимости донора, преимущественно братьев или сестер больного, лучше близнецов.
АНЕМИЯ ГЕМОЛИТИЧЕСКАЯ НАСЛЕДСТВЕННАЯ МИКРОСФЕРОЦИТАРНАЯ (синонимы: семейная гемолитическая желтуха, болезнь Минковского— Шоффара). Одна из самых распространенных в нашей стране форм малокровия, возникающего вследствие повышенного разрушения эритроцитов и сопровождающегося желтухой.
Этиология и патогенез. В основе заболевания лежит аномалия эритроцитов, характеризующихся неправильной формой (сфероцитоз). Такие эритроцитыподвергаются усиленному разрушению преимущественно в селезенке (внутриклеточный гемолиз), вследствие чего развиваются анемия, гемолитическая желтуха, гиперплазия селезенки.
Клиническая картина. Характерны иктеричность кожных покровов и слизистых оболочек, спленомегалия, реже — увеличение печени, иногда изменения костей черепа: «башенный череп», широкая переносица, высокое небо.
Диагноз. Устанавливают на основании клинико-гематологической картины и семейного анамнеза (симптомы гемолитической желтухи у членов семьи).
Прогноз. Чаще благоприятный. Смерть в период острого тяжелого гемолитического криза — явление довольно редкое. В этих случаях прогноз зависит от срока спленэктомии.
Лечение. Радикальным методом лечения является удаление селезенки — основного органа кроверазрушения, после чего наступает клиническое выздоровление, хотя сфероцитоз и сниженная осмотическая резистентность эритроцитов остаются. Антианемические мероприятия (гемотран-сфузии цельной крови, применение витамина В12, железа и кортикостероидов) малоэффективны и даже противопоказаны.
АНЕМИЯ ГЕМОЛИТИЧЕСКАЯ ПРИОБРЕТЕННАЯ характеризуется желтухой и анемией вследствие внутрисосудистого гемолиза.
Этиология и патогенез. Под влиянием физических, химических, лекарственных средств, бактериальных, инфекционно-токсических, паразитарных вырабатываются антиэритроцитарные аутоантитела, приводящие к усиленному гемолизу эритроцитов.
Клиническая картина. Нарушение общего состояния, слабость, головные боли, желтуха различной интенсивности, субфебрильная температура тела, умеренное увеличение селезенки (чаще у детей младшего возраста), иногда печени. В крови — признаки анемии различной степени. Течение хроническое, однако могут наблюдаться тяжелые гемолитические кризы с высокой температурой тела, признаками глубокой анемии, гемоглобинурией.
Диагноз. Устанавливают на основании клинико-гематологической картины, отсутствия выраженного сфероцитоза и положительной пробы Кумбса (прямой или непрямой).
Прогноз. Менее благоприятен, чем при семейной гемолитической желтухе. Возможны рецидивы.
Критерием выздоровления служит появление отрицательной пробы Кумбса.
Лечение. Применяют кортикостероидные гормоны (преднизолон по 0,5—1,0 мг/кг), особенно при гемолитических кризах, трансфузии одногруппной индивидуально совместимой эритроцитной массы, подобранной по отрицательной непрямой пробе Кумбса.
АНЕМИЯ ИНФЕКЦИОННАЯ И ПОСТИНФЕКЦИОННАЯ. Малокровие, развивающееся у детей в результате инфекционного заболевания.
Этиология и патогенез. Причины анемии этого типа: грипп, ангина, пневмония, скарлатина, дифтерия, брюшной тиф, паратифы, ревматизм, туберкулез, бруцеллез, сепсис. У детей младшего возраста — отит (особенно рецидивирующий), пиелонефрит, энтероколит. Анемия усугубляется авитаминозами, сопровождающими инфекционный процесс.
Клиническая картина. Общими симптомами являются слабость, вялость, плохой аппетит, температурная реакция, бледность кожи и слизистых оболочек.
Диагноз. Основывается на анамнезе и клинико-гематологической картине.
Прогноз. В значительной степени зависит от основного заболевания. После ликвидации инфекционного процесса в большинстве случаев благоприятный.
Лечение. Терапия основного заболевания. При гипохромии применяют препараты железа, витаминотерапию.
АНЕМИЯ ПОСТГЕМОРРАГИЧЕСКАЯ (острая и хроническая). Малокровие, возникающее вследствие обильных однократных или незначительных, но длительно повторяющихся кровотечений.
Этиология и патогенез. Малокровие новорожденных может быть обусловлено повреждением сосудов плаценты или пуповины и появлением как наружного, так и скрытого кровотечения. Кровопотери у детей грудного, младшего и старшего возраста бывают связаны с механической травмой, геморрагическими диатезами, заболеваниями органов кроветворения, желудочно-кишечного тракта (варикозное расширение вен пищевода, полипы), почек (гломерулонефрит).
Острая потеря 30—33 % от общего количества крови может привести к шоку со смертельным исходом. При хроническом кровотечении организм ребенка может перенести большую кровопотерю (до 50 %).
Клиническая картина. Характерны слабость, головокружения, бледность кожных покровов и видимых слизистых оболочек; тахикардия, систолический шум на верхушке сердца. В крови — признаки нормо- или гипохромной анемии, анизоцитоз, небольшой лейкоцитоз, умеренный ретикулоцитоз.
Диагноз. Не представляет трудностей.
Прогноз. Зависит от остроты кровопотери и характера болезни, сопровождающейся кровотечением.
Лечение. Остановка локального кровотечения. Трансфузия цельной свежей крови с гемостатической целью, эритроцитной массы в качестве заместительной терапии, тромбоплазмы; аскорбиновая кислота, рутин. Препараты железа; диета, богатая белками, витаминами, лечение основного заболевания.
ВАСКУЛИТ ГЕМОРРАГИЧЕСКИЙ (синонимы: болезнь Шенлейна—Геноха, геморрагический капилляро-токсикоз). Заболевание сосудистой системы (микро-тромбоваскулит), характеризующееся полиморфной сыпью и волнообразным течение. Этиология и патогенез. Болезнь возникает под действием: различных вирусных и бактериальных инфекций, прививок, травм, охлаждения, лекарственных и пищевых аллергенов. В основе ее лежат повреждающее действие циркулирующих иммунных комплексов на стенки микрососудов, нарушение микроциркуляции, микротромбоз.
Клиническая картина. Различают в основном кожно-суставную и абдоминальную формы заболевания. Характерны общее недомогание, бледность, пастозность, повышение температуры тела от субфебрильных до высоких цифр; полиморфная, симметрично расположенная папулезно-геморрагическая сыпь, нередко явления полиартрита (припухлость, болезненность суставов).При абдоминальном синдроме отмечаются резкие приступообразные боли в животе, симулирующие картину острого живота, обусловленные геморрагическими высыпаниями на брюшине и стенках кишечника, симптомы кишечного кровотечения.Одновременно наблюдаются и кожные высыпания. Поражения почек, протекающие по типу подострого гломерулонефрита, утяжеляют состояние ребенка и ухудшают прогноз. Изменений периферической крови нет. Количество тромбоцитов нормальное. Длительность кровотечения не изменена.
Диагноз. При характерной клинической картине несложен.
Прогноз. В большинстве случаев благоприятный. Значительно ухудшается при почечном синдроме в связи с возможностью развития хронического гломерулонефрита, а также при инвагинации или перфорации кишечника.
Лечение. Строгий постельный режим. Гепаринотерапия (100— 300 ЕД/кг в сутки подкожно) под строгим лабораторным контролем. Назначают криоплазму, курантил, десенсибилизирующие средства (тавегил, супрастин, димедрол по 0,015—0,025 г 2 раза в день), аскорбиновую кислоту, рутин, витамин Р, в тяжелых случаях — преднизолон по 5—15 мг в сутки. Диета с исключением индивидуальных сенсибилизирующих факторов.
ПУРПУРА ТРОМБОЦИТОПЕНИЧЕСКАЯ (синонимы: геморрагическая тромбоцитопения, болезнь Верльгофа, идиопатическая тромбоцитопени-ческая пурпура — ИТП). Форма геморрагического диатеза, вызванного уменьшением количества тромбоцитов в крови и снижением резистентности капилляров.
Этиология и патогенез. Наиболее часто встречаются иммунные формы ИТП. Под влиянием различных эндо- и экзогенных факторов образуются антитромбоцитарные антитела (иммуноглобулины класса С, реже М), фиксирующиеся на поверхности тромбоцитов и вызывающие усиленное их разрушение.
часто встречаются при кори, краснухе, скарлатине, брюшном тифе, вирусном гепатите, грипп), сепсисе, эндокардите, хронической тонзиллогенной интоксикации, тиреотоксикозе, болезнях системы крови, а также при воздействии химических, лучевых, лекарственных (стрептомицин, барбитураты) факторов.
Клиническая картина. Характерны несимметрично расположенные полиморфные геморрагии от мелких петехий до обширных подкожных кровоизлияний, возникающих спонтанно или под влиянием малейшей травмы; кровоточивость слизистных оболочек рта, носа, желудочно-кишечного тракта, иногда легочные, почечные, маточные кровотечения. Селезенка, как правило, не увеличена. Полостные кровоизлияния наблюдаются редко. Симптом Кончаловского («жгута») положителен. Основной гематологический признак — тромбоцитопения, иногда отмечаются умеренный лейкоцитоз, нейтрофилез. Показатели красной крови зависят от степени кровопотери.Длительность кровотечения увеличена (по Дюке).
Диагноз. Ставят на основании характерной клинической картины, тромбоцитопении.
Прогноз. Относительно благоприятный. С возрастом наклонность к кровоточивости уменьшается. Смертельную опасность могут представлять профузные кровотечения и кровоизлияния в жизненно важные органы.
Лечение. Назначают дицинон, аскорбиновую кислоту (до 1 г в сутки), витамин Р, кортикостероидные гормоны (преднизолон по 1—2 мг/кг в сутки в течение 1,5—2 мес), гемостатические мероприятия при кровотечениях: трансфузии свежей плазмы по 30— 50 мг, тромбоцитной взвеси по 150—200 мл.
При маточных кровотечениях применяютжидкий экстракт водяного перца. При отсутствии стойкого эффекта от энергичной консервативной терапии (с применением кортикостероидов), при непрекращающихся профузных кровотечениях и угрозе кровоизлияния в мозг показана спленэктомия.
ГЕМОФИЛИЯ. Форма кровоточивости, свойственная лицам мужского пола, выявляющаяся в раннем детстве.
Этиология и патогенез. Заболевание семейно-наследственного характера, связанное с нарушением системы гемостаза в первой фазе. Кровоточивость при гемофилии вызвана дефицитом фактора VIII — антигемофильного глобулина (гемофилия А), фактора IX — Кристмаса (гемофилия В) или некоторых других (гемофилия С и др.).
К л и н и ч е с к а я картина.Характерны кровоизлияния в ткани при малейшей травме; длительные, иногда профузные кровотечения после экстракции зубов; частые кровоизлияния в суставы (гемартрозы), анкилозы (блок сустава, нарушение подвижности) носовые, горловые, почечные кровотечения. При обширных гематомах повышается температура тела, отмечается небольшая билирубинемия.После обильных кровотечений развивается постгеморрагическая анемия. Количество тромбоцитов, ретракция сгустка, длительность кровотечения не изменены.
Диагноз. Основывается на клинической картине и семейном анамнезе. Дифференциация типов гемофилии возможна только при специальном исследовании свертывающей системы крови.
Прогноз. Серьезен в связи с возможностью профузных кровотечений и кровоизлияний в жизненно важные органы.
Лечение.Тщательный уход, профилактика травм, ушибов. При обострении применяют систематические трансфузии свежей антигемофильной плазмы (по 50—100 мл) и антигемофильного глобулина, криопреципитата; при кровотечениях — местные гемостатические мероприятия (тампонада, давящая повязка, холодные компрессы), повторные (через 6—8 ч) переливания свежей плазмы. Хлорид кальция, викасол малоэффективны. Больным гемофилией В показано переливание консервированной крови. При гемартрозах показаны покой, холодные компрессы. Тяжелые анкилозы требуют ортопедического лечения (этапная редрессация с помощью гипсовых повязок). Профилактика рецидивов проводится путем периодического введения криопреципитата.
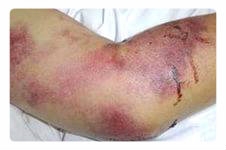 Гемофилия (haemophilia) – наследственная патология из группы коагулопатий, приводящая к нарушению синтеза VIII, IX либо XI факторов, отвечающих за свертываемость крови, заканчивающееся его недостаточностью. Заболевание характеризуется повышенной склонностью как к спонтанным, так и имеющим причины геморрагиям: внутрибрюшинным и внутримышечным гематомам, внутрисуставным (гемартрозам), кровотечениям пищеварительного тракта, неспособности свертывания крови при различных даже незначительных травмах кожных покровов.
Гемофилия (haemophilia) – наследственная патология из группы коагулопатий, приводящая к нарушению синтеза VIII, IX либо XI факторов, отвечающих за свертываемость крови, заканчивающееся его недостаточностью. Заболевание характеризуется повышенной склонностью как к спонтанным, так и имеющим причины геморрагиям: внутрибрюшинным и внутримышечным гематомам, внутрисуставным (гемартрозам), кровотечениям пищеварительного тракта, неспособности свертывания крови при различных даже незначительных травмах кожных покровов.
Болезнь актуальна в педиатрии, так как выявляется у детей младшего возраста, чаще в первый год жизни младенца.
История появления гемофилии уходит глубоко в старину. В те времена она была широко распространена в обществе, особенно в царских семья как Европы, так и России. Целые династии венценосных особ мужского пола страдали от гемофилии. Отсюда произошли термины «гемофилия венценосных» и «царская болезнь».
Примеры хорошо известны – гемофилией болела английская королева Виктория, передавшая ее по наследству своим потомкам. Ее же праправнуком был русский царевич Алексей Николаевич, сын императора Николая II, получивший в наследство «царскую болезнь».
Этиология и генетика
Причины болезни связаны с мутациями в гене, который сцеплен с X-хромосомой. В результате этого отсутствует антигемофилический глобулин и появляется дефицит ряда других плазменных факторов, входящих в состав активного тромбопластина.
Гемофилия имеет рецессивный тип наследования, то есть передается по женской линии, но заболевают ей только мужчины. У женщин тоже имеется поврежденный ген, но они не болеют, а выступают только как его носитель, передавая патологию своим сыновьям.

Появление здорового или больного потомства зависит от генотипа родителей. Если в браке муж здоров, а жена является носителем, то у них с вероятностью 50/50 могут родиться как здоровые, так и больные гемофилией сыновья. А у дочерей есть 50% шансов заполучить дефектный ген. У мужчины, страдающего заболеванием и имеющего генотип с мутированным геном, и здоровой женщины появляются на свет дочери-носители гена и совершенно здоровые сыновья. Девочки с врожденной гемофилией могут появиться от матери-носителя и больного отца. Такие случаи очень редки, но все же встречаются.
Наследственная гемофилия выявляется в 70% случаев от общего числа больных, на оставшиеся 30% приходится обнаружение спорадических форм заболевания, связанных с мутацией в локусе. В последующем подобная спонтанная форма становится наследственной.
Классификация
Код гемофилии по МКБ-10 – D 66.0, D67.0, D68.1
Типы гемофилии различаются в зависимости от недостатка того или иного фактора, способствующего гемостазу:
• Гемофилия типа А (классическая). Характеризуется рецессивной мутацией гена F8 в половой Х-хромосоме. Это наиболее распространенный тип заболевания, встречающийся у 85% больных, характеризуется врожденным дефицитом антигемофильного глобулина, приводящего к сбою образования активной тромбокиназы.
• Болезнь Кристмаса или гемофилия типа В связана с недостаточностью IX фактора, по-другому называемого фактором Кристмаса – плазменного компонента тромбопластина, тоже участвующего в формировании тромбокиназы. Такой тип болезни выявляется не более чем в у 13% пациентов.
• Болезнь Розенталя или гемофилия типа С (приобретенная) отличается аутосомно-рецессивным или доминантным типом наследования. При этом типе дефектен фактор XI. Диагностируется лишь у 1–2% от общего числа больных.
• Конкомитированная гемофилия – очень редко встречающаяся форма с одновременным недостатком VIII и IX факторов.
Гемофилия типа А и В обнаруживается исключительно у мужчин, типа С – у обоих полов.
Остальные разновидности, например, гипопроконвертинемия, очень редки, на них приходится не более 0,5% от всех пациентов с гемофилией.
Клинические проявления
Тяжесть кинического течения не зависит от типа болезни, а определяется уровнем дефицитного антигемофильного фактора. Различают несколько форм:
• Легкую, характеризующуюся уровнем фактора от 5 до 15%. Дебют заболевания обычно приходится на школьные годы, в редких случаях после 20 лет, и связан с оперативным вмешательством либо травмами. Кровотечения редкие и неинтенсивный.
• Средней тяжести. С концентрацией антигемофильного фактора до 6% от нормы. Появляется в дошкольном возрасте в виде умеренно выраженного геморрагического синдрома, обостряющегося до 3 раз в год.
• Тяжелая выставляется при концентрации недостающего фактора до 3% от нормы. Сопровождается выраженным геморрагическим синдромом с раннего детского возраста. У новорожденного младенца отмечается длительное кровотечение из пуповины, мелена, кефалогематомы. При развитии ребенка – посттравматические или спонтанные кровоизлияния в мышцы, внутренние органы, суставы. Возможны длительные по времени кровотечения от прорезывания или смены молочных зубов.
• Скрытая (латентная) форма. С показателем фактора, превышающим 15% от нормы.
• Субклиническая. Антигемофильный фактор не снижается ниже 16–35%.
У маленьких детей кровотечения могут возникать от прикусывания губы, щеки, языка. После перенесенных инфекций (ветрянки, гриппа, ОРВИ, кори) возможны обострения геморрагического диатеза. Ввиду частых и продолжительных кровотечений обнаруживается тромбоцитопения и анемия разного вида и степени тяжести.
Характерные признаки гемофилии:
• Гемартрозы – обильные кровотечения в суставы. По чистоте кровоизлияний на них приходится от 70 до 80%. Чаще страдают голеностопные, локтевые, коленные, реже – тазобедренные, плечевые и мелкие суставы пальцев рук и ног. После первых кровоизлияний в синовиальные капсулы кровь постепенно рассасывается без каких-либо осложнений, функция сустава полностью восстанавливается. Повторные кровотечения приводят к неполному рассасыванию, образованию фибринозных сгустков, откладываемых в капсуле сустава и хряще с постепенным их прорастанием соединительной тканью. Проявляется выраженными болями и ограничением движения в суставе. Рецидивирующие гемартрозы становятся причиной облитерации, анкилозы суставов, гемофилического остеоартроза и хронического синовита.
• Кровотечения в костную ткань заканчиваются декальцинации костей и асептическому некрозу.
• Кровоизлияния в мышцы и подкожную клетчатку (от 10 до 20%). Излившаяся в мышцы или межмышечные пространства кровь долгое время не сворачивается, поэтому без труда проникает в фасции и близлежащее ткани. Клиника подкожных и внутримышечных гематом – плохо рассасываемые синяки разных размеров. Как осложнения возможны гангрены либо параличи, появляющиеся вследствие сдавления объемными гематомами крупных артерий или периферических нервных стволов. Это сопровождается выраженным болевым синдромом.
Статистика
На территории России насчитывается около 15 тысяч мужчин, больных гемофилией, из них детей примерно 6 тысяч. В мире же проживает более 400 тысяч человек с таким заболеванием.
• Длительные кровотечения из слизистых десен, носа, полости рта, разных отделов желудка либо кишечника, а также из почек. Частота появления до 8% от общего числа всех кровотечений. Любые медицинские манипуляции или операции, будь то удаление зуба, тонзиллэктомия, внутримышечный укол или прививка, заканчиваются обильным и длительным кровотечением. Крайне опасны кровотечения из слизистой гортани и зева, так как это может закончиться обструкцией дыхательных путей.
• Кровоизлияния в разные отделы головного мозга и мозговые оболочки приводят к нарушениям нервной системы и соответствующей симптоматике, нередко заканчиваясь смертью больного.
• Гематурия самопроизвольная либо из-за травм поясничного отдела. Обнаруживается в 15–20% случаев. Симптомы и нарушения, ей предшествующие – расстройства мочеиспускания, пиелонефрит, гидронефроз, пиелоэктазия. Больные обращают внимание на появление крови в моче.
Для геморрагического синдрома характерно отсроченное появление кровотечения. В зависимости от интенсивности травмы оно может возникнуть спустя 6–12 часов и позднее.
Приобретенная гемофилия сопровождается нарушением цветового восприятия (дальтонизм). Встречается в детском возрасте редко, только лишь при миелопролиферативных и аутоиммунных заболеваниях, когда начинают вырабатываться антитела к факторам. Только у 40% пациентов удается выявить причины появления приобретенной гемофилии, к ним относят беременность, болезни аутоиммунного характера, прием некоторых медикаментов, злокачественные новообразования.
При появлении вышеперечисленных проявлений человек должен обратиться в специализированный центр по лечению гемофилии, где ему назначат обследование и при необходимости лечение.
Диагностика
На этапе планирования беременности, будущие родители могут пройти медико-генетическую консультацию с молекулярно-генетическим исследованием и сбором генеалогических данных.
Перинатальная диагностика заключается в проведении амниоцентеза или биопсии хорина с последующим ДНК-исследованием полученного клеточного материала.
Диагноз устанавливается после проведения детального обследования и дифференциальной диагностики пациента.
Обязательно физикальное обследование с осмотром, аускультацией, пальпацией, сбором семейного анамнеза для выявления возможного наследования.
Лабораторные исследования гемостаза:
- коагулограмма;
- количественное определение факторов IX и VIII;
- определение МНО – международного нормализованного отношения;
- анализ крови для подсчета количества фибриногена;
- тромбоэластография;
- тромбодинамика;
- протромбиновый индекс;
- подсчет АЧТВ (активированное частичное тромбопластиновое время).
Появление у человека гемартроза требует рентгенографии пораженного сустава, а гематурии – дополнительного исследования мочи и функции почек. Ультразвуковая диагностика проводится при забрюшинных кровоизлияниях и гематомах в фасции внутренних органов. При подозрении на кровоизлияния в мозг обязательно назначают КТ или МРТ.
Дифференциальную диагностику поводят с тромбастенией Глянцманна, пурпурой тромбоцитопенической, болезнью Виллебранда, тромбоцитопатией.
Лечение
Заболевание неизлечимо, но поддается заместительной гемостатической терапии концентратами недостающих факторов. Доза концентрата подбирается в зависимости от степени его дефицита, выраженности гемофилии, вида и тяжести кровотечения.
Важно начать лечение при первых кровотечениях. Это помогает избежать многих осложнений, требующих уже хирургического вмешательства.

Лечение состоит из двух составляющих – постоянного поддерживающего или профилактического и немедленного, при проявлениях геморрагического синдрома. Поддерживающее лечение заключается в периодическом самостоятельном внутривенном введении концентрата антигемофильного фактора. Задача врачей – предотвратить появление артропатий и кровотечения в различные части тела. При тяжелой форме гемофилии частота введения доходит до 2–3 раз в неделю при профилактическом лечении и до 2 раз в день при основном.
Основа лечения – антигемофильные препараты, переливания крови и ее компонентов.
• Гемостатическая терапия гемофилии типа А подразумевает использование криопреципитата – концентрата антигемофильного глобулина, изготовленного из свежезамороженной плазмы человека.
• Гемофилию типа В лечат в/в введением PPSB – комплексного препарата, в состав которого входят несколько факторов, в том числе протромбин, проконвертин и компонент тромбопластина плазмы. В дополнение вводят свежезамороженную плазму донора.
• При гемофилии типа С используют свежезамороженную сухую плазму.
Симптоматическое лечение состоит из назначения глюкокортикоидов, ангиопротекторов. Дополняется физиотерапией. Первая помощь при внешних кровотечениях включает местное прикладывание гемостатической губки, обработку раны тромбином, наложение временной давящей повязки.
Вследствие интенсивной заместительно-трансфузионной терапии возникает ингибиторная форма гемофилии, характеризующаяся появлением ингибиторов к факторам свертываемости, которые нейтрализуют вводимый больному антигемофильный фактор, приводя к бесполезности лечения. Положение спасает плазмаферез и назначение иммунодепрессантов.
При кровоизлиянии в сустав рекомендован покой в течение 3–5 дней, нестероидные противовоспалительные средства в таблетках и глюкокортикоиды местно. Хирургическое лечение показано при необратимом нарушении функции сустава, его разрушении.
Альтернативное лечение
В дополнении к медикаментозному лечению больные могут лечиться средствами народной медицины. Профилактику кровотечений можно проводить с помощью трав, обладающих вяжущим свойством, способствующим укреплению стенок сосудов. К ним относят тысячелистник, экстракт виноградных косточек, чернику, крапиву двудомную.
Для улучшения свертывания крови принимают следующие лекарственные растения: арнику, кориандр, астрагал, корень одуванчика, плоды софоры японской и другие.
Осложнения
Осложнения делятся на группы.
• Связанные с геморрагиями:
а) кишечная непроходимость или сдавление мочеточников обширными гематомами;
б) деформации опорно-двигательного аппарата – гипотрофия мышц, узурирование хряща, искривление таза или позвоночного столба, как осложнение гемофильного остеоартроза;
в) инфицирование гематом;
г) абструкция дыхательных путей.
• Связанные с иммунной системой – появление ингибиторов факторов, затрудняющих лечение.
Больнее находятся в зоне риска заражения ВИЧ-инфекцией, герпетической и цитомегаловирусной инфекцией и вирусными гепатитами.
Профилактика
Специфической профилактики не существует. Возможна лишь медикаментозная профилактика, предотвращающая возникновение кровотечений. При вступлении в брак и планировании беременности важно пройти медико-генетическое консультирование со всем необходимым обследованием.
Прогноз
При легкой форме прогноз благоприятный. При тяжелой – значительно ухудшается. В целом зависит от вида, степени тяжести, своевременности лечения и его эффективности. Больного ставят на учет, дают инвалидность.
Сколько живут при разных видах гемофилии? Легкая форма не влияет на продолжительность жизни больного. Эффективное и постоянное лечение при среднетяжелой и тяжелой форме помогает жить больному столько же, сколько живут здоровые. Смерть наступает в большинстве случаев после кровоизлияний в головной мозг.
ЛЕЙКОЗ ОСТРЫЙ. С истемное заболевание крови опухолевой природы с нарушением созревания кроветворных клеток на ранних стадиях развития.
Этиология и патогенез. Причина заболевания окончательно не выяснена. Канцерогенным эффектом обладают химические (бензол.) и физические (ионизирующая радиация) факторы. В происхождении лейкозов играют роль гормональные, иммунные нарушения.
Лейкоз характеризуется:
а) гиперплазией кроветворной ткани;
б) метаплазией — появление патологических очагов кроветворения в костном мозге и различных органах;
в) клеточной анаплазией — утрата родоначальными клетками кроветворения способности к дифференцировке.
Поражение сосудов, глубокая тромбоцитопения, агранулоцитоз, иммунные нарушения влекут за собой ряд тяжелых осложнений (геморрагии, инфаркты, язвенно-некротические изменения, присоединение инфекций).
Клиническая картина. Начало лейкозов не имеет строгой клинической картины. Изменения со стороны верхних дыхательных путей, ангина, пневмония, боли в костях и суставах, беспричинный субфебрилитет или высокая лихорадка, системная лимфаденопатия, увеличение размеров печени и селезенки — все эти явления при острых лейкозах можно встретить в качестве первых симптомов. В крови — признаки анемии, тромбоцитопения, лейкопения или лейкоцитоз, значительно увеличенная СОЭ. В лейкограмме — гранулоцитопения, большое количество недифференцированных клеток, лимфобластов, миелобластов.
Различают:
1. начальную стадию заболевания
2. период развернутых клинико-гематологических проявлений
3. стадию ремиссии
4. рецидива заболевания (костномозгового, нейролейкоз, специфическое поражение половых желез)
5. терминальную стадию заболевания.
Диагноз. Основывается на клинико-гематологической картине. Особое значение для диагностики имеет пункция костного мозга, (морфологическое, цитохимическое, имуноцитологическое исследования бластных клеток).
Прогноз. Современная терапия с соблюдением всех ее принципов позволяет у 85—90 % больных добиться полной ремиссии. О практическом выздоровлении от острого лимфобластного лейкоза можно говорить после 6—7-летнего безрецидивного течения заболевания.
Лечение. Состоит из следующих этапов:
1. Проведение 4— 6 курсов полихимиотерапии: введение метотрексата в комбинации с цитозаром.
2. Закрепление ремиссии. Наступление полной ремиссии обязательно должно подтверждаться контрольным исследованием костного мозга. Проводится закрепляющий курс полихимиотерапии.
3. Поддерживающее лечение последовательным (каждые 1,5—2 мес) чередованием 6-меркаптопурина и метотрексата с применением циклофосфана 1 раз в 7— 10 дней.
4. Эффективность терапии улучшается при использовании иммунокорриги-рующей терапии.
ЛЕЙКОЗ ХРОНИЧЕСКИЙ. Системный гиперпластический процесс с нарушением созревания клеток гранулоцитарного рада на стадии промиелоцитов, миелоцитов.
Этиология и патогенез. Причина неизвестна. Патогенез хронического миелолейкоза, как и острого лейкоза, заключается в гиперплазии кроветворной ткани, развитии очагов миелоидной метаплазии в лимфатических узлах, печени, селезенке, почках и других органах. Клиническая картина.
У детей выделяют следующие клинические стадии заболевания:
I стадия — начальная, доклиническая;
II стадия развернутых клинических проявлений;
III стадия — переходный период;
IV стадия — острая фаза, «бластный» криз,
V терминальный перио