Жидкофазное окисление органических веществ молекулярным кислородом можно ускорить соединениями металлов переменной валентности. Ускорение обусловлено каталитическим распадом гидропероксидов под действием ионов металлов переменной валентности, в результате чего возникают свободные радикалы, инициирующие окисление. Ионы металлов переменной валентности вступают с гидропероксидом в окислительно-восстановительные реакции (цикл Габера - Вейса), в результате чего резко возрастает скорость реакций окисления.
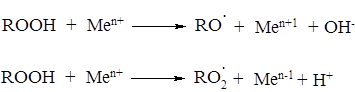
Реакции гидропероксида с металлом предшествует, часто, комплексообразование:
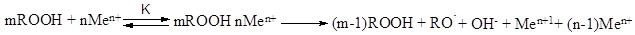
Чаще всего образуются комплексы состава 1:1, так что реакция протекает как бимолекулярная со скоростью V i = k [ROOH]m[Me n+ ]n. При высокой концентрации ROOH, когда все катионы катализатора связаны в комплексе с гидропероксидом, реакция имеет первый порядок по катализатору и нулевой по гидропероксиду, а в общем случае скорость распада ROOH:
VROOH =2 k [ROOH][Men+1](1+ K [ROOH])-1
Соединения таких металлов, как Co, Mn, Ce вызывают гомолитический распад гидропероксидов с выходом на распавшийся ROOH, близким к 100%. Однако, соединения других металлов переменной валентности, например, Cr, V, Mo, разрушают ROOH с очень низким выходом радикалов. Очевидно, эти катализаторы ведут распад ROOH по двум направлениям: гетеролитическому (основной) (k m) и гомолитическому (побочный) (ki). Соотношение между гомолитическим и гетеролитическим направлениями зависит от металла и лигандного окружения. Если преобладает распад ROOH на молекулярные продукты (k m >> ki), то такие соединения металлов переменной валентности не будут ускорять автоокисление.
В окисляющемся углеводороде ROOH – не единственный окислитель, в нем образуются такие активные окислители, как пероксидные радикалы. Они также вступают в реакцию с соединениями металлов. Это приводит к тому, что в каталитическом окислении углеводородов только часть RO2· превращается в гидропероксид, а часть RO2· по реакции с катализатором превращается в спирт или кетон. Способность соединений переходных металлов быстро реагировать как с RO2·, так и с ROOH приводит к их двойственной функции при окислении углеводородов. Когда соль металла переменной валентности (Co, Mn, Ce, Cu, Fe) вводится в углеводород, где уже есть гидропероксид, то наблюдается ускоренное протекание реакции, т. е. катализ, обусловленный гомолитическим разрушением гидропероксида и инициирующим действием системы: катализатор - гидропероксид. Если соль металла переменной валентности катализатор вводится в углеводород, не содержащий ROOH, то в эффект ускорения как правило отсутствует, а в некоторых случаях наблюдается тормозящее действие такого соединения. Это объясняется тем, что возникающие в системе пероксильные радикалы разрушаются, реагируя с металлом переменной валентности, и такой обрыв цепей сдерживает цепной процесс окисления, пока не образуется гидропероксид в количестве, достаточном для инициирования окисления.
В биологических системах ионы металлов переходной группы также инициируют окислительные реакции. Инициирование происходит за счет образования комплексных соединений с кислородом. Это обусловлено:
1) Перекрывание в таких комплексах внешних орбиталей иона металла и кислорода ведет к трансформации характера реакций окисления органических субстратов со спин-запрещенных в спин-разрешенные. Более того, если молекула кислорода вступает в несимметричное взаимодействие с ионом металла, то энергетическое вырождение ее разрыхляющих орбиталей снимается и термодинамически выгодным может оказаться (как это имеет место при фиксации кислорода в поле лигандов гема) спаривание обоих внешних электронов на одной разрыхляющей орбитале:
__ (расщепление уровней О2
↑ ↑ ↑↓ __ в поле лигандов металла-
О2 1О2 ↑↓ О2 комплексообразователя)
Действительно, в большинстве случаев комплексы кислорода с переходными металлами диамагнитны, что прямо указывает на снятие в таких комплексах вырождения π-разрыхляющих орбиталей О2, а расщепление их уровней становится настолько большим, что оба электрона переходят на одну из этих орбиталей (на ту или иную - в зависимости от геометрии образующегося комплексного соединения).
Как правило, молекулярный кислород достаточно эффективно взаимодействует лишь с ионами металлов, находящихся в низких степенях окисления: например, с Fe2+ или Cu+, но не c Fe3+ или Cu2+. Даже если комплексы Fe3+ и Cu2+ являются ненасыщенными (то есть, когда они обладают пустой координационной позицией, доступной для связывания соответствующего лиганда), связывания О2 не происходит. Это не удивительно, так как "хорошими" лигандами для высокоокисленных ионов металлов являются обычно относительно сильные основания, к которым молекулярный кислород не принадлежит.
2) Поскольку суммарный заряд комплекса металл-кислород является положительным, то такой комплекс значительно более «охотно» акцептирует электроны при окислении субстратов, чем нейтральная молекула кислорода, что также приводит к усилению активности молекулы кислорода.
В результате взаимодействия О2 с низкоокисленными ионами металлов переходной группы, последние, помимо описанной выше активации этой молекулы, выступают также и донорами электронов для О2, что благоприятствует дальнейшей активации кислорода для осуществления им более эффективного окисления органических субстратов. При одноэлектронном восстановлении кислорода даже некомплексированными ионами металлов эта реакция термодинамически выгодна:
Fe2+ + O2 ® Fe3+ + O2- (ΔGo = -128.9 кДж/моль)
Cu+ + O2 ® Cu2+ + O2- (ΔGo = -69.1 кДж/моль)
(в качестве примера изображены наиболее биологически значимые ионы металлов).
Типичный путь активации молекулярного кислорода неспецифически комплексированными ионами металлов, осуществляемый через серию нескольких редокс-этапов, можно изобразить следующим образом (где Мn - ион металла переходной группы в низкой степени окисления, L - лиганд, LM - комплексное соединение):
LMn + O2 ® LM(n+1)-O2-
LM(n+1)-O2- ® LM(n+1) + O2-
2O2- + 2H+ ® H2O2 + O2
LMn + O2- ® LM(n+1)-O22-25)
LMn + H2O2 ® LMn-O22- (26)
Именно образующиеся металл-перекисные интермедиаты и являются ответственными за дальнейшую продукцию наиболее мощных из так называемых «активных форм кислорода» (АФК), которые в дальнейшем выступают объектом нашего рассмотрения.
Общепризнанный механизм металл-комплексной активации пероксид-лиганда может включать как гомолитический распад перекиси на свободные радикалы (реакция Фентона):
LMn + H2O2 ® LMn-O22-(Н2) ® LM(n+1) + OH- + HO·,
так и осуществляться путем гетеролитического расщепления связи О-О:
LMn + H2O2 ® LMn-O22-(Н2) ® LM(n+2)=O + H2O
В результате двухэлектронного восстановления ионами металла-комплексообразователя продуцируются металл-оксокомплексы высоких степеней окисления, которые действуют уже аналогично гидроксильному радикалу (НО·) в отношении реакций отрыва атома водорода или электрона:
LM(n+2)=O + H2O +e- ® LM(n+1)(OH) + OH-
По сути металлоксокомплексы - это координационные соединения, у которых оксид-анион (О2-) является лигандом, непосредственно связанным с металлом-комплексообразователем.
В свободном состоянии ион О2-, так же как и другие многозарядные анионы, существовать не может. Если для атома кислорода сродство к электрону составляет около 142 кДж/моль, то между ионом О- и электроном возникает электростатическое отталкивание: на кривой изменения потенциальной энергии системы О- + е- = О2- потенциальной ямы нет (ни одно положение е- вблизи О- не может быть устойчиво). Нейтрализация же заряда у О- протоном ведет к образованию мощнейшего акцептора е- - гидроксильного радикала, НО·.
Формально эти структуры можно представить себе как результат депротонирования молекулы воды (выступающей в качестве лиганда в аквакомплексах):
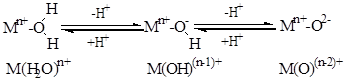
Кроме того, эти электрондефицитные субстанции, наряду с отрывом электронов у субстрата-восстановителя, способны выступать и в качестве поставщиков атома кислорода при их взаимодействии с органическими молекулами.
Забегая далеко вперед, отметим, что у пероксидаз - ферментов, утилизирующих наиболее устойчивые к окислению органические молекулы и других субстанции (например, галоидные ионы), в качестве активного центра активных форм таких выступает оксоферрильная группировка: FeIV=O. Очевидно, что этим обстоятельством и определяется исключительно высокая окислительная способность таких ферментов (Е0³+1.0 В), играющих ведущую роль как в уничтожении инфицирующих микроорганизмов лейкоцитами, так и на биосферном уровне - в разложении микроорганизмами лигнина.
Оба рассмотренных выше пути металл-зависимой активации молекулы О2 (гомо- и гетеролитический), в случае наиболее распространенного in vivo металла - железа, ведут к образованию высоко активных Fe-оксосоединений, отличающихся только степенью окисления; но лишь гомолитический механизм продуцирует НО·-радикалы:
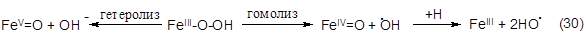
Аналогичные процессы, по-видимому, реализуются и в активных центрах ферментов, утилизирующих молекулярный кислород для осуществления оксидазных и оксигеназных реакций в живых системах. В ходе этих реакций промежуточные токсические интермедиаты восстановления кислорода не выделяются в окружающее пространство, а подвергаются превращению до конечных продуктов Н2О2 и Н2О.