Синдром Марфана
| Синдром Марфана - системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. | 
|
В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки.
Симптомы: синдром Марфана характеризуется сочетанным поражением скелета, глаз, сердечно-сосудистой и нервной систем.
Клиника: больные отличаются высоким ростом, относительно коротким туловищем с непропорционально длинными тонкими конечностями (долихостеномелией) и удлиненными паукообразными пальцами (арахнодактилией), астеническим телосложением со слаборазвитой подкожной клетчаткой и мышечной гипотонией, длинным и узким лицевым скелетом (долихоцефалией), наличием высокого аркообразного неба и нарушения прикуса (прогнатии).
Отмечаются нарушение функции суставов (гипермобильность), деформация грудной клетки (воронкообразная или килевидная форма), деформация позвоночника (сколиоз, кифоз, кифосколиоз, подвывихи и вывихи шейного отдела, спондилолистез), а также плсокостопия и протрузия вертлужной впадины.
Изменения аорты у больных синдромом Марфана характеризуются прогрессирующим расширением ее восходящей части и клапанного кольца (дилатацией, аннулоаортальной эктазией) и аневризмами; поражение митрального клапана - миксоматозной дегенерацией створок, патологическим удлинением и разрывом створочных хорд, обызвествлением клапанного кольца. У плода с синдромом Марфана возможно формирование врожденных пороков сердца - коарктации аорты, стеноза легочной артерии, ДМПП и ДМЖП. Органические и функциональные изменения сердца и сосудов у больных синдромом Марфана часто сопровождаются нарушением ритма (наджелудочковой и желудочковой тахикардией, фибрилляцией предсердий) и развитием инфекционного эндокардита.
Самая неблагоприятная неонатальная форма синдрома Марфана проявляется в классическом варианте уже при рождении, приводит к прогрессирующей сердечной недостаточности и летальному исходу на первом году жизни ребенка.
Для большинства случаев синдрома Марфана характерна патология органа зрения, включающая близорукость, вывих и подвывих (эктопию) хрусталика, уплощение и увеличение размера роговицы, гипоплазию радужной оболочки и цилиарной мышцы, косоглазие, изменение калибра сосудов сетчатки.
При синдроме Марфана наблюдается поражение других систем и органов: нервной (эктазия твердой мозговой оболочки, пояснично-крестцовое менингоцеле), бронхолегочной (спонтанный пневмоторакс, эмфизема легких, дыхательная недостаточность), кожи и мягких тканей (атрофические стрии), рецидивирующие паховыеи бедренные грыжи, вывихи и разрывы связок, а также эктопия почек, опущение мочевого пузыря и матки, варикозное расширение вен и др.
Характерный для синдрома Марфана выброс адреналина может способствовать постоянному нервному возбуждению, гиперактивности, а иногда развитию неординарных способностей и умственной одарённсоти.
Лечение: синдром Марфана неизлечим. Этиотропной терапии недуга не существует, ведь невозможно заменить гены ребенка. Больным проводят симптоматическую терапию, целью которой является облегчение общего состояния, устранение симптомов и предупреждение тяжелых осложнений.
Прогерия
| Прогерия «преждевременное старение»– это редкое генетическое заболевание, впервые описанное Гилфордом, которое проявляется преждевременным старением организма, связанное с его недоразвитием. Прогерия классифицируется на детскую- синдром Гетчинсона (Хатчинсона)-Гилфорда и взрослую – синдром Вернера. |

|
Симптомы: у детей замедляется рост, увеличиваются размеры черепа, но лицевая часть остается маленькой, нижняя челюсть – недоразвитой. Формируется клювовидный тонкий нос и экзофтальм (выпячивание глаз). Тотально выпадают волосы – на голове и теле, ресницы и брови. При алопеции разрушаются волосяные фолликулы, рост волос становится невозможным. На волосистой части головы выбухают вены. Дерма и подкожная клетчатка подвергаются атрофическим изменениям: кожа истончается, иссушается, покрывается морщинами.
На открытых участках кожи появляются пигментные пятна. Ногти недоразвитые, ломкие, утолщенные, округло выпуклы. К 2 годам развивается фиброз органов и околосуставных тканей. Пассивные движения в локтевых и коленных суставах ограничены, формируется характерное положение костей ног – «поза всадника». Скелет подвергается гипоплазии, дисплазии и дегенеративным изменениям. К 5-6 годам диагностируется атеросклероз сосудов, сердечные шумы, гипертрофия левого желудочка, помутнение хрусталика, резистентность к инсулину. Половые органы остаются недоразвитыми. Уровень умственного развития обычно выше, чем у сверстников.
Клинические проявления синдрома Вернера обнаруживаются с 14 до 18 лет. Подросток начинает отставать в росте, его волосы седеют и выпадают. Кожа лица и конечностей бледнеет, истончается, натягивается. Мышечная и жировая ткани атрофируются, руки и ноги становятся непропорционально тонкими, кожа над суставными выступами изъязвляется. К 30 годам появляется катаракта, голос слабеет и хрипнет, на ногах образуются язвы, на подошвах – мозоли, сосудистые звездочки, кератоз. Внешний вид пациентов специфичен: низкий рост, лунообразное лицо, выступающий подбородок, суженное ротовое отверстие.
Сальные и потовые железы атрофируются. Костно-суставные изменения включают метастатическую кальцификацию, явления генерализованного остеопороза, эрозивных остеоартритов, ограниченную подвижность и деформацию пальцев рук, сгибательные контрактуры, боли в конечностях, артрит и остеомиелит. Отмечаются атеросклеротические изменения сосудов, медленно прогрессирует катаракта, снижаются интеллектуальные способности. Причиной смерти становится онкопатология или тяжелое сердечно-сосудистое заболевание.
Диагноз устанавливается на основании клинико-анамнестических данных.
Опрос. Уточняют время появления симптомов, их выраженность, степень физической и социальной дезадаптации, наличие периода обычного развития (6-24 месяца при детской форме, 14-20 лет – при взрослой). Собирают данные семейного и генеалогического анамнеза, выявляя генетическую патологию или ее отсутствие в роду.
Осмотр. Специалисты оценивают неврологический статус, пассивную и активную подвижность суставов, костные деформации, состояние кожи и подкожной клетчатки, сохранность зрения, интеллектуальных функций. Инструментальные исследования: рентгенография костей, УЗИ и МР-томография органов, ЭКГ, офтальмоскопия.
Биогенетическое исследование. Выполняется анализ генетического материала методом секвенирования ДНК. Исследуются участки генов, дефекты в которых приводят к развитию прогерии. У пациентов с детской и взрослой формой болезни обнаруживаются мутации в гомозиготном и компаунд-гетерозиготном состоянии.
Дифференциальный диагноз: исключают системную склеродермию, пойкилодермию, синдром Ротмунда-Томсона, синдром Видемана-Раутенштрауха.
Лечение: специфические методы терапии прогерии не разработаны. Медицинская помощь пациентам заключается в облегчении симптомов болезни. Проводится лечение атеросклероза, остеопороза, остеомиелита, катаракты, диабета, патологий сердца и онкологических заболеваний.
Синдром Олбрайта
| Синдром Олбрайта - генетическое заболевание невыясненной этиологии, поражающее весь организм человека. Патология возникает сравнительно редко и проявляется асимметричной гиперпигментацией кожи, гормональным дисбалансом, полиостотической фиброзной дисплазией костей. | 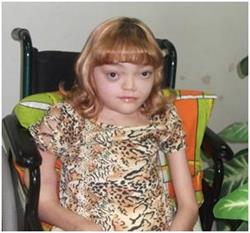
|
Синдром Олбрайта развивается в основном у представительниц прекрасной половины человечества. Он обусловлен спонтанной мутацией гена в процессе эмбриогенеза и не передается по наследству. Диагноз ставят детям возрасте 5-10 лет на основании признаков, характерных для данного заболевания.
Впервые клиника синдрома была описана в 1907 году ученым Олбрайтом, в честь которого он и получил свое название. Ученый наблюдал за низкорослыми и коренастыми девочками с круглым лицом и короткой шеей. У них были обнаружены мышечные спазмы, скелетные деформации, частичное отсутствие зубной эмали, отставание в умственном развитии, признаки ожирения. Эндокринопатия проявлялась ранним половым созреванием: появлением менструаций, активным ростом грудных желез, появлением волос на лобке. Происхождение патологии Олбрайт объяснял резистентностью почечного эпителия к паратгормону, приводящей к ускоренному обратному всасыванию фосфора и развитию гиперфосфатемии. При этом у больных паращитовидные железы имели нормальное строение, а гуморальные изменения отсутствовали. В официальной медицине синдром получил название псевдогипопаратиреоз. Нарушения фосфорно-кальциевого обмена приводят к патологии костной системы. Подобные расстройства легко объяснить устойчивостью тканей к эндогенному паратгормону. У больных отмечается отставание в психофизическом развитии. При этом отсутствует реакция на введение экзогенного паратгормона.
Больным с синдромом Олбрайта требуется консультация специалистов в области гинекологии, эндокринологии, лучевой диагностики, офтальмологии, травматологии и ортопедии.
Симптомы: больных развивается фиброзная остеодисплазия, проявляющаяся различными деформациями и скелетной кривизной. Чаще всего поражаются трубчатые кости на одной конечности, что приводит к хромоте и деформации позвоночника. Уже в раннем возрасте нарушается формирование скелета. У пациентов выявляется деформация челюсти и черепа: лицевые кости утолщаются, становятся широкими, грубыми и крупными. Так формируются внешние уродства: асимметричное лицо и односторонний экзофтальм. Затем появляются симптомы артрита. Фиброзно-кистозная дисплазия представляет собой фокальное замещение кортикального слоя кости пролиферирующими фибробластами. Образование кист приводит к деформации кости и возникновению переломов. Чаще всего встречается деформация бедра по типу «пастушьего посоха». На коже больных появляются участки гиперпигментации, представляющие собой скопления пятен неправильной формы с неравными краями. Окраска пятен часто варьируется от желтоватой до темно-коричневой. Очаги поражения локализуются на ногах, бедрах, туловище. Пятна имеют достаточно большие размеры и гладкую поверхность. Они присутствуют на коже с рождения или распространяются по мере роста ребенка. Эпидермис при этом не изменяется. Их интенсивный рост требует гистологического исследования и оперативного вмешательства.
Клиника: у больных наблюдаются судороги, которые могу возникать как сами по себе, так и после воздействия раздражающих факторов (нервное напряжение, перепад температур, тяжелая физическая работа и т.д.). Кальцинаты, располагающиеся в коже и подкожно-жировой клетчатке, могут изъязвляться и доставлять дискомфорт пациентам. При этом больной будет обращаться за помощью не к эндокринологу или генетику, а к хирургу или дерматологу. Такие люди, как правило, низкого роста, у них круглое одутловатое лицо, ожирение и укорочение пальцев. Наблюдаются ложные суставы там, где их быть не должно, отсутствует подвижность в тех местах, где она предполагается анатомически, нарушение конфигурации суставов и костей. Среди симптомов также рвота, кровь в моче, катаракта и изменения в зубной эмали. Кроме того, из-за низкого уровня кальция в крови с детства у пациентов наблюдается снижение интеллекта и задержка развития.
Лечение: синдром Олбрайта — неизлечимое заболевание. Для поддержания жизни больных на оптимальном уровне и устранения основных клинических признаков болезни проводят симптоматическую терапию. Если общетерапевтические мероприятия не будут проведены вовремя, задержка умственного развития только усугубится.
Ихтиоз
| Ихтиоз – это наследственное заболевание кожи, протекающее по типу дерматоза. Характеризуется диффузным нарушением ороговения и проявляется в виде чешуек на коже, которые напоминают рыбью чешую. | 
|
Ихтиоз передается по наследству. В более редких случаях у человека может возникнуть приобретенный ихтиоз. Заболевание не является следствием инфицирования, а это значит, что заразиться им нельзя.
Первые признаки ихтиоза у больных появляются еще в детском возрасте, а иногда даже сразу после рождения. Люди, страдающие наследственным ихтиозом, часто страдают заболеваниями щитовидной железы, репродуктивных органов, а также надпочечников. Нередко у них присутствует дефицит иммунитета (клеточного и гуморального).
У этих пациентов практически всегда нарушена работа потовых желез, что в сочетании с дефицитом витамина A может привести к усиленному ороговению кожных покровов.
При данной патологии в кожных покровах всегда присутствует избыточное количество кератина, структура которого нарушена. При этом отторжение старых клеток кожи происходит чрезвычайно медленно. В результате этого на коже человека появляются чешуйки, в пространстве между которыми собираются аминокислотные комплексы, провоцирующие их затвердение. По этой же причине чешуйки крепко соединены друг с другом.
У пациентов, страдающих ихтиозом, отмечаются сухие и ломкие волосы и ногти, у многих зубы поражены кариесом.
При ихтиозе часто наблюдаются заболевания глаз: такие как ретинит, конъюнктивит и близорукость.
Виды ихтиоза:
вульгарный ихтиоз;
ламеллярный ихтиоз;
Х-сцепленный ихтиоз;
болезнь Дарье;
ихтиозиформная эритродермия.
Симптомы: сухость кожи, шелушение, покраснение кожи (эритродермия), деформация ногтей, истончение и ломкость волос, пиодермия (гнойно-воспалительное заболевание кожи), конъюнктивит, выраженный кожный рисунок на ладонях и стопах.
Клиника: вульгарный ихтиоз - наиболее выражено шелушение на разгибательных поверхностях конечностей, меньше поражены кожа спины и живота, волосистой части головы. На коже голеней чешуйки самые темные и толстые, полигональной формы, плотно прикрепленные. Фолликулярный гиперкератоз в виде мелких суховатых узелков в устьях волосяных фолликулов наблюдается на коже бедер, плеч, предплечий и ягодиц, также может локализоваться на коже туловища, лица. Ладони и подошвы имеют подчеркнутый рисунок, повышенную складчатость, что придает им старческий вид. Ногтевые пластинки ломкие, крошатся со свободного края, иногда развивается онихолизис. Волосы истончаются, становятся редкими.
Ихтиоз, связанный с Х-хромосомой - сразу после рождения отмечается сухость кожного покрова. Позже появляются светло- и темно-коричневые чешуйки на разгибательных поверхностях конечностей. Задняя поверхность шеи из-за скопления чешуек приобретает «грязный» вид. Подмышечные впадины, локтевые ямки и область гениталий свободны от поражений. Отличительной особенностью от других форм ихтиоза является отсутствие поражения кожи лица и кистей по типу «перчаток».
Пластинчатый ихтиоз - отмечается генерализованное пластинчатое шелушение, одновременно с которым обязательно наблюдается ладонно-подошвенный гиперкератоз, являющийся постоянным клиническим признаком заболевания. Чешуйки на гладкой коже обычно мелкие и светлые, на голенях крупные, образуют пластинчатое шелушение. У некоторых больных наблюдается деформация ушных раковин.
Врожденная буллезная ихтиозиформная эритродермия - в области крупных естественных складок (коленных, локтевых, лучезапястных и голеностопных суставов, на шейных складках, в области подмышечных впадин) наблюдается гиперкератоз с крупнопластинчатыми роговыми крошкоподобными образованиями. Очаги гиперкератоза бурого, буро-черного или грязно-серого цвета. На фоне гиперкератоза первоначально появляются пузыри с серозным содержимым, в последующем присоединяется вторичная инфекция. Одновременно отмечается повышение температуры тела и увеличение регионарных лимфатических узлов
Ихтиоз плода - на голове больного наблюдается толстый слой роговых наслоений, имеющиеся волосы короткие, редкие или вовсе отсутствуют. Лицо деформировано и покрыто крупными роговыми пластинами. Рот широко раскрыт из-за сильной инфильтрации мягких тканей, в углах рта выявляются глубокие трещины. Губы утолщены, их слизистая оболочка вывернута, наблюдается резко выраженный эктропион и разряженность ресниц. Ушные раковины деформированы и плотно прижаты к черепу или завернуты вперед. В ноздрях и слуховых проходах выявляются роговые наслоения в виде пробок.
Лечение: снижение сухости кожных покровов и размягчение роговых чешуек (увлажняющие кремы и мази; кератолитические средства; ретиноиды).
Синдром «кошачьего крика»
| Синдром «кошачьего крика» – хромосомное нарушение, обусловленное делецией (отсутствием) фрагмента короткого плеча 5-ой хромосомы. | 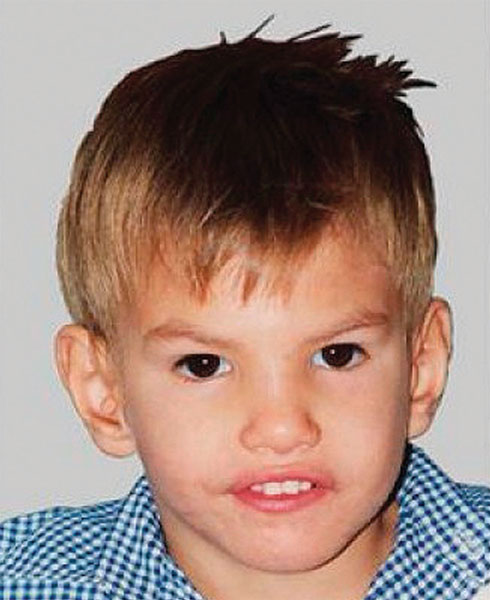
|
Плач новорожденных с синдромом «кошачьего крика» по звуку напоминает кошачье мяуканье, что и послужило названию патологии. Кроме этого, у детей имеет место микроцефалия, лунообразное лицо, косоглазие, аномалии прикуса, различные врожденные пороки, грубое интеллектуальное недоразвитие и т. д. Синдром «кошачьего крика» диагностируется на основании совокупности характерных признаков и цитогенетического исследования.
Симптомы: Новорождённые рождаются, как правило, доношенными, но с небольшой пренатальной гипотрофией. Беременность у матери может протекать абсолютно нормально или сопровождаться угрозой самопроизвольного прерывания не чаще, чем в популяции. Наиболее патогномоничным ранним признаком синдрома является плач ребенка, который напоминает мяуканье кошки. Высокое и пронзительное звучание детского крика обусловлено анатомическими особенностями строения гортани при данном синдроме – узостью ее просвета, небольшим надгортанником, необычной складчатостью слизистой оболочки, мягкой консистенцией хрящей. Некоторые авторы считают, что специфический крик имеет центральное происхождение и не связан с недоразвитием гортани.
Клиника: основными клиническими симптомами данной патологии, кроме характерного плача, напоминающего крик кошки, являются: нарушение сосания и глотания, замедленное физическое развитие, задержка речевого и умственного развития, избыточное слюноотделение, частые запоры и нетипичные черты лица. Главной причиной развития многих симптомов, в том числе и появления характерного крика, связано с вовлечением в патологический процесс нервной системы и структурных элементов гортани. К дополнительным клиническим признакам, которые отмечаются при данной патологии, относятся микроцефалия, гипотония, полные щёки на круглом лице, эпикантус, косоглазие, низко посаженные уши, пороки сердца, короткие пальцы. Достаточно редко данная патология проявляется расщеплением нёба или верхней губы, дисплазией тимуса, мегаколоном, врождённым вывихом бедра, кишечной непроходимостью, крипторхизмом, пороками развития почек, гипоспадией, плоскостопием, сращение пальцев рук и ног, повышенной гибкостью суставов и различными дерматоглифическими признаками.
Лечение: специфического лечения данного хромосомного заболевания в настоящее время не существует. Для стимуляции психомоторного развития под наблюдением детского невролога проводятся курсы медикаментозной терапии, массажа, физиотерапии, ЛФК. Дети с синдромом «кошачьего крика» нуждаются в помощи психологов, дефектологов, логопедов. Врожденные пороки сердца при синдроме «кошачьего крика» часто требуют хирургической коррекции, поэтому детям необходима консультация кардиохирурга, проведение Эхо, ЭКГ и других необходимых исследований. Дети с патологией мочевыделительной системы должны находиться под наблюдением детского нефролога и периодически проходить комплекс необходимых обследований (УЗИ почек, общий анализ мочи, биохимическое исследование крови и мочи и др.).