 | |||||
 | |||||
 | |||||
 |  | ||||||
|
| ||||||
Гипоперфу- Нарушение Нарушение



 зия тканей потребления использования
зия тканей потребления использования

 кислорода глюкозы
кислорода глюкозы






|
|
| ||||||
 |
Существенный вклад в нарушение метаболизма вносят медиаторные системы (биологически активные вещества): гистамин, серотонин, кинины, простагландины, лизосомальные ферменты, комплемент, интерлейкины. При длительных и выраженных расстройствах микроциркуляции страдают так называемые «шоковые» органы – легкие, почки, печень, сердце, мозг, форменные элементы крови – эритроциты и тромбоциты. Преимущественное поражение того или иного шокового органа определяет дальнейшую картину шока. Так, поражение легких ведет к дыхательной недостаточности (нарушения вентиляции, диффузии, перфузии), почек – почечной недостаточности (анурии, олигурии, снижение экскреции креатинина, продуктов азотистого обмена, нарушению концентрационной функции почек и т. п.). Поражение печени сопровождается расстройством метаболической, дезинтоксикационной, гомеостатической и других функций этого важного органа.
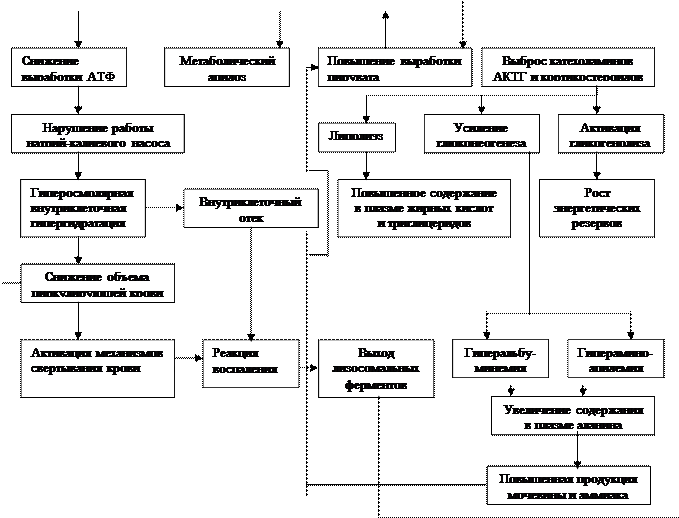
Схема 3
Схема основных механизмов синдрома
Мультисистемной полиорганной недостаточности
 | |||||
 | |||||
 |
Массивный системный
 |  | ||||||||
 | |||||||||
 | |||||||||
 |
В патогенезе развития шока существеннейшее значение приобретают так называемые «порочные круги», когда первоначальные нарушения деятельности одних органов и систем могут потенцироваться расстройствами других. Так, функциональные изменения в ЦНС сопровождаются выраженными дыхательными и гемодинамическими сдвигами, которые, в свою очередь, отягощают гипоксические состояния жизненно важных органов, усугубляя нарушения нервных процессов в самой центральной нервной системе.
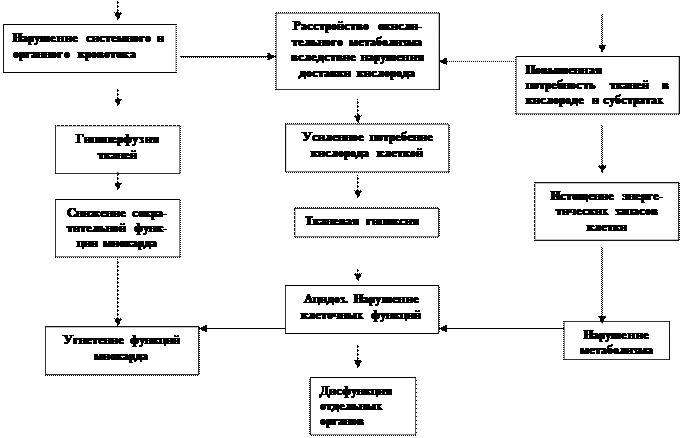
Клеточное звено. Заключительный этап нарушений функций организма при любом виде шока, в том числе и травматическом, обусловлен расстройствами клеточного метаболизма (Схемы 2 и 3). Они связаны с ограничением поступления в клетку адекватного количества кислорода или невозможностью использовать его клеткой. Вследствие дефицита кислорода тканевое дыхание сменяется гликолизом, энергетическая ценность которого не может полностью обеспечить клетку потребным количеством АТФ. Кроме того, гликолиз способствует развитию лактоацидоза и метаболического ацидоза.
Большое значение в развитии метаболических расстройств имеет повреждение цитоплазматических и внутриклеточных мембран, вызванное усилением свободнорадикальных процессов и перекисного окисления липидных компонентов биомембран. Без АТФ цитоплазматическая мембрана теряет способность поддерживать важнейшие градиенты, в первую очередь, электрохимический градиент по натрию и калию (расстройство функции натрий-калиевого насоса). Цитоплазма наводняется ионами натрия, хлора, водой, теряя калий. Это ведет к повышению возбудимости нервных и мышечных клеток, в частности кардиомиоцитов, что сопровождается дальнейшими глубокими, порой необратимыми изменениями со стороны сердечной мышцы (и ЦНС).
Изменения электрофизиологических характеристик миокарда сопровождается нарушениями автоматии и сократительной функции сердца, что ведет к дальнейшим снижению МОС и ограничению перфузии тканей. Снижение сократительной функции миокарда обусловлено также повышением содержания в крови особого фактора, угнетающего сократительную способность миокарда при шоке.
Поступившая вследствие нарушения проницаемости цитоплазматической мембраны из межклеточных пространств вода, с одной стороны, вызывает внутриклеточный отек, с другой – уменьшает общий объем циркулирующий в организме жидкости. Внутриклеточная дисгидрия разрушает лизосомы клеток, и лизосомальные ферменты попадают в общий кровоток и околоклеточные пространства, где оказывают свое литическое действие на соседние клетки.
Появление в крови фракций лизосомальных ферментов запускает процессы внутрисосудистого свертывания крови, активирует комплемент и калликреин-кининовую систему, а нарастающий ацидоз продолжает усугублять функции ферментных систем клеток.
Нарушения использования глюкозы. Они могут возникать вследствие ограниченного поступления глюкозы в клетку или в результате расстройства ее утилизации. Неадекватное снабжение клетки глюкозой может быть связано с гиповолемией, ишемией и другими патологическими нарушениями системного, периферического и микроциркуляторного русла. Если шок сопровождается септическими процессами, то метаболизм глюкозы осложняется лихорадкой, бактериемией, наличием вазоактивных токсинов, эндотоксинов и других БАВ.
Кроме того, целый ряд компенсаторных механизмов, активируемых шоком, снижает потребление глюкозы клетками. Среди них называют кортикостероиды, соматотропный гормон, катехоламины, повышенную резистентность к инсулину, тахикардию, усиление сократимости миокарда, повышенный МОС. Для выживания в этих условиях в клетке усиливаются процессы гликогенолиза, глюконеогенеза, липолиза, что в конечном итоге ведет к истощению запасов углеводов и жиров и развитию клеточной недостаточности.
Существенный вклад в развитие клеточной недостаточности вносит расстройство метаболизма белков, которое выражается в распаде протеинов первоначально до аминокислот (асептический аутоканнибализм), затем до мочевины, мочевой кислоты, аммиака, которые наводняют кровь, токсически действуя на клетку.
При длительных и выраженных расстройствах микроциркуляции в первую очередь страдают так называемые «шоковые органы» – легкие, сердце, почки, печень, мозг, форменные элементы крови – эритроциты и тромбоциты. Как указывалось, преимущественное поражение того или иного шокового органа и определяет дальнейшую картину шока.
Небезынтересно отметить, что, по Кулагину выделяют три фазы шока: (1) нервную; (2) сосудистую; (3) метаболическую.
Терминальная стадия шока характеризуется полным истощением механизмов компенсации, выраженными метаболическими, циркуляторными сдвигами, прогрессирующими нарушениями функций органов и систем, дистрофическими и некротическими изменениями в паренхиматозных органах, например печени («шоковая печень»). Расстройства кровообращения на терминальной стадии шока не корригируются.
Таким образом, основным патогенетическим звеном поздних стадий шока является циркуляторная гипоксия, обусловленная гипоперфузией тканей и нарушением микроциркуляции, приводящая к развитию разветвленных цепей метаболических изменений как патологического, так и защитно-приспособительного характера.
Наконец, следует остановиться и на классификации шока по степени его тяжести, в основу которого положены показатели величины систолического АД, степень кровопотери и тяжесть состояния:
I степень (АД до 100 мм рт.ст., кровопотеря до 1 л);
II степень (АД до 80 мм рт.ст., кровопотеря - 1-1,5 л);
III степень (АД 60 мм рт.ст., кровопотеря 1,5-2 л);
IV степень – терминальные состояния (АД не определяется): преагония, агония, клиническая смерть. Критическим уровнем АД при шоке следует считать величину в 70 мм рт.ст.
Коллапс
Другой формой острой гипотензии является коллапс. Коллапс – это остро развивающаяся форма сосудистой недостаточности, которая характеризуется первичным падением сосудистого тонуса, а также острым уменьшением объема циркулирующей крови. В результате этого снижается приток венозной крови к сердцу, уменьшается ударный и далее минутный объем сердца, падает артериальное и венозное давление, нарушается перфузия тканей и, соответственно, обмен веществ, формируется гипоксия и ацидоз, в первую очередь головного мозга с угнетением жизненно-важных центров и органов.
Этиология. В интересах клиники выделяют в зависимости от этиологических факторов следующие формы коллапса: чаще всего коллапс развивается при интоксикациях и острых инфекционных заболеваниях, который получил наименование (1) токсический коллапс (острые отравления цианидами, нитратами, окисью углерода и т.п.); (2) коллапс вследствие действия физических факторов (электрический ток, ионизирующая радиация, действие высоких температур и т.п.); (3) коллапс при эндогенных интоксикациях и инфекционных болезнях (перитонит, аллергия и т.п.); (4) гипоксический коллапс (в условиях пониженного парциального давления кислорода, особенно в сочетании с гипобарией и гипокапнией); (5) ортостатический коллапс – переход из горизонтального положения в вертикальное; (6) коллапс при кессонной болезни; (7) геморрагический коллапс; (8) коллапс вследствие заболеваний сердца.
Патогенез. Развитие коллапса следует связать с двумя основными механизмами. Первый заключается в падении тонуса артериол, венул и вен в результате воздействия тех факторов, которые вызвали коллапс (физические, инфекционно-токсические, аллергические и прочие) непосредственно на (1) сосудодвигательный центр; (2) сосудистую стенку; (3) рецепторы сосудистых рефлексогенных зон (синокаротидная, аортальная и другие). Это ведет к снижению периферического сопротивления, и при недостаточности компенсаторных механизмов, регулирующих периферическое сопротивление, наступает парез сосудов, что ведет к патологическому увеличению емкости сосудистого русла, уменьшению объема циркулирующей крови с депонированием ее в определенных сосудистых регионах, ограничению венозного притока крови к сердцу и быстро развивающемуся падению АД.
Второй механизм развития коллапса связан с быстрым уменьшением массы циркулирующей крови (например, при кровотечении, потере плазмы и других биологических жидкостей). Возникающий в ответ на это рефлекторный спазм мелких сосудов, а также учащение сокращений сердца под влиянием усиленного выброса катехоламинов могут оказаться недостаточными для сохранения нормального уровня АД.
Уменьшение объема циркулирующей крови снижает венозный возврат ее к сердцу по венам большого круга кровообращения и соответственно сердечный выброс, нарушает систему микроциркуляции. Это ведет к скоплению крови в капиллярах и падению АД. Развиваются гипоксия циркуляторного типа, ацидоз и другие метаболические нарушения. Гипоксия и ацидоз приводят к повышению проницаемости сосудистой стенки. Потеря тонуса прекапиллярных сфинктеров и ослабление их чувствительности к вазопрессорным веществам развиваются на фоне сохранения тонуса посткапиллярных сфинктеров, которые более устойчивы к ацидозу.
При повышении проницаемости стенок капилляров наблюдается переход воды и электролитов из крови в межклеточные пространства. Нарушаются реологические свойства крови, развивается гиперкоагуляция и патологическая агрегация эритроцитов и тромбоцитов, создаются благоприятные условия для формирования микротромбов.
Таким образом, видно, что коллапс имеет много общего с шоком, и американские медики не дифференцируют шок и коллапс. Однако у нас придерживаются нескольких иных принципиальных позиций, отличающих шок от коллапса:
1. Коллапс сопровождается потерей сознания, а шок нет;
2. В развитии коллапса нет смены последовательных фаз нарушений сосудистого тонуса, в то время как шок является фазовым процессом (эректильная, торпидная, терминальная фазы);
3. При формировании коллапса тонус сосудов изменяется первично, а во время шока – вторично.
Следующей формой острой сосудистой недостаточности является обморок. Обморок – это самая легкая форма сосудистой недостаточности. Обморок, или синкопальное состояние (syncope) – это внезапно развивающееся патологическое состояние, которое характеризуется временной потерей сознания вследствие анемизации головного мозга и вегето-сосудистыми расстройствами.
Этиология и патогенез. Причинами обморока могут быть различные факторы, вызывающие преходящий спазм сосудов головного мозга, в результате чего развивается глубокая гипоксия или возникает ограничение утилизации кислорода, что также расценивается как гипоксия. В большинстве случаев обморок имеет рефлекторный нейрогенный генез. Обморок могут вызвать: (1) отрицательные эмоции (испуг, неприятное зрелище, конфликтная ситуация и т.п.); (2) боль; (3) применение лекарственных препаратов (например, ганглиоблокаторов); (4) раздражение некоторых рецепторных зон (например, синокаротидной, области блуждающего нерва – вазовагальный обморок и т.д.); (5) нарушения адаптационных механизмов; (6) чрезмерные физические нагрузки; (7) перемена положения тела; (8) многочисленные заболевания крови, сердечнососудистой системы, гипогликемические состояния, нарушение кислородтранспортной функции организма.
Клинически обморок проявляется тремя стадиями:
1. Предобморочное состояние;
2. Нарушение сознания;
3. Восстановительный период.
.
4. Нет стадийности.
АТЕРОСКЛЕРОЗ
Атеросклерозпредставляет собойхроническое прогрессирующее заболевание эластических и мышечно-эластических артерий, характеризующееся пролиферативно-синтетическим ответом гладкомышечных клеток и фибробластов сосудистой стенки, моноцитов-макрофагов и тромбоцитов на патологические липопротеиды с формированием в интиме фиброзно-липидных бляшек – атером. Дальнейшая эволюция атером сопровождается реактивными изменениями медии, приводящими к типичным осложнениям – изъязвлениям, кальцинозу, тромбозу и эмболиям, аневризмам и кровотечениям. Атеромы преобразуются в источник вазоконстрикторных БАВ. Характер реакций пораженного сосуда на вазомоторные стимулы извращается – доминирует тенденция к спазму, затрудняется вазодилатация, в результате чего развиваются ишемические поражения органов.
Атеросклероз как специфическую разновидность артериосклероза, при котором происходит отложение липидов в сосудистой стенке артерий эластического типа, впервые выделил Ф. Маршан (1904). Само наименование процесса происходит от греческих слов «кашица» и «твёрдый». Артериосклероз – более широкое понятие, известное с 1833 г. и включающее все процессы, при которых происходят фиброз и сужение артериальных сосудов любого калибра. Помимо атеросклероза, в эту категорию включают артериосклероз Мёнкеберга и артериолосклероз (гиалиновую и гиперпластическую разновидности). В отличие от атеросклероза, эти процессы первично охватывают медию, наблюдаются в средних и малых артериях мышечного типа (артериосклероз Мёнкеберга) или даже в артериолах (артериолосклероз), а также не связаны с накоплением липопротеидов.
Таким образом, отличия атеросклероза от других видов артериосклероза существенны, хотя эти процессы в средних артериях могут комбинироваться у одного и того же пациента. Ясно, что атеросклероз имеет наибольшее эпидемиологическое значение и представляется уникальным среди данных процессов, т.к. он этиологически прямо связан с нарушением липидного обмена.
Эпидемиология и медицинская география атеросклероза. Атеросклероз – одна из главнейших медицинских проблем ХХ столетия, поскольку в Европе и Северной Америке само заболевание, его прямые последствия и осложнения служат ведущей причиной смертности населения. На протяжении последних 70 лет в развитых странах отмечалось прогрессирующее учащение и отягощение проявлений атеросклероза и их «омоложение». Так, в США с 1930 по 1970 годы смертность от атеросклероза коронарных артерий возросла в 40 раз. В 70-80 годы наивысший показатель заболеваемости атеросклерозом наблюдалась у населения стран Западной Европы – Англии и соседних с ней Шотландии, Уэльса, Ирландии, Исландии, Дании, Финляндии, Швеции, а также Австралии и Новой Зеландии, Северной Америки, ряде государств Азии, включая Россию (особенно, северо-западную). Везде смертность от ИБС регистрировалась между 200 и 300 на 100000 населения. С 1968 года в Северной Америке наметилась стойкая, удерживающаяся в последние 30 лет тенденция к понижению смертности от атеросклероза, которая для США в 1990 году упала с 242 до 102 на 100000 населения. В России, Великобритании, Скандинавии и многих других стран и поныне эпидемиологическая картина остается практически столь же тревожной – более половины людей умирает именно в результате ИБС. Весьма низкая пораженность атеросклерозом наблюдается в странах Африки, Азии (кроме Японии, где она на уровне Южной Европы). В то же время, инсульт, особенно, ишемический, в Японии как раз встречается в 4-5 раз чаще, чем в Финляндии и США.
Прямыми последствиями атеросклероза являются:
1. Ишемическая болезнь сердца (ИБС).
2. Ишемическая болезнь мозга (инсульт, ишемическая энцефалопатия).
3. Ишемическое заболевание конечностей (гангрена нижних конечностей, перемежающаяся хромота и тромбоз подвздошных артерий).
4. Ишемическая болезнь кишечника, обусловливающая атонические состояния и другие расстройства функций желудочно-кишечного тракта у пожилых пациентов.
5. Атеросклероз почечных артерий (первично сморщенная почка, хроническая почечная недостаточность).
Итак, атеросклероз чаще всего встречается у белого городского населения Северной и Средней Европы и Северной Америки. Он поражает и негроидов, но сравнительно редок среди монголоидов. В происхождении межпопуляционных различий по частоте атеросклероза и его осложнений роль климата минимальна. Эскимосы живут севернее, чем датчане и шведы, но имеют очень низкую частоту ИБС. Значительно больше пораженность этой болезнью зависит, по-видимому, от особенностей питания и образа жизни населения, социально-экономических факторов. Показана прямая корреляция между уровнем потребления холестерина, насыщенных жиров и сахара и частотой атеросклероза. Зарегистрировано обратное соотношение между количеством в диете непредельных жиров (особенно, содержащих ненасыщенные жирные кислоты), пищевых волокон, антиоксидантов и смертностью от ИБС. Однако, и эти закономерности не абсолютны – восточноафриканское племя масаев использует диету с рекордно высоким содержанием холестерина (до 1,5 г/сутки), а атеросклероз у этих скотоводов и охотников саванны даже в глубокой старости находится лишь на начальных стадиях, причем ИБС у них практически не встречается. Роль образа жизни доказывается тем, что у мигрантов – выходцев из регионов с низкой распространенностью атеросклероза, например, японцев и арабов, при переселении в США поражение атеросклерозом увеличивается и приближается к характерной для аборигенов.
Не столь существенна роль социальных факторов. В СССР и США в конце 70-х годов влияние уровня образования на частоту инфаркта миокарда у мужчин, жителей мегаполисов, было противоположным. В цитадели империализма интеллигенция оказалась несколько менее подверженной инфарктам, чем неквалифицированные рабочие. Но в стране победившего пролетариата тенденция была обратной.
Очевидно, огромное значение имеет разная распространенность патологических аллелей генов апопротеинов и неодинаковая частота наследственных гиперлипопротеинемий у населения разных регионов. Так, ген липопротеида(а) наиболее распространен именно в высоко пораженных атеросклерозом скандинавских популяциях. Семейная наследственная гиперхолестеринемия очень часто встречается среди англосаксов и кельтских народов (до 2% носителей дефектного гена), но практически отсутствует среди монголов, что коррелирует с распространенностью ИБС, являющейся главной причиной смертности представителей англосаксонских этносов, но казуистически редкой среди потомков Чингисхана.
Особую доказательность имеют исследования частоты атеросклероза у представителей разных этнических общин, проживающих на одной территории. Красноречивым примером может служить заболеваемость инфарктом миокарда в городе-государстве Сингапур. Несмотря на идентичность климатогеографических и экологических факторов, сингапурские индусы поражаются инфарктом в 10 раз чаще местных китайцев и в 8 раз чаще местных малайцев. Можно заключить, что атеросклероз – мультифакториальное заболевание, при котором играет роль действие комплекса факторов, связанных с питанием, образом жизни, экологическими условиями и, в первую очередь, генетическими особенностями организма. Как отмечал Ю.А. Лопухин, «Ничто – ни диета, ни строжайший контроль за факторами риска, ни лекарственные средства, ни бег трусцой, ни специальные комплексы физических упражнений – не в состоянии изменить широкое распространение атеросклероза, роста инфарктов сердца и мозговых инсультов – главных могильщиков наиболее деятельной и продуктивной части современного населения».
Этиология атеросклероза. Модели и факторы риска. Вскоре после идентификации атеросклероза Ф. Маршаном удалось идентифицировать липиды, которые были ответственны за развитие атероматозных бляшек. Ими оказались липопротеиды, содержание главным образом холестерин и его эфиры.
Напомним, что синтез холестерина осуществляется в печеночных клетках из ацетата. Кроме того, часть холестерина поступает в организм из пищи. У человека ежесуточно синтезируется около 800 мг холестерина и около 400 мг всасывается из кишечника. У здоровых людей натощак содержание холестерина и триглицеридов в крови – довольно постоянные величины (данные Таблицы 6).
В крови холестерин циркулирует в виде макромолекулярных компонентов – липопротеидов, имеющих различную плотность. Самое большое количество холестерина содержат липопротеиды низкой плотности (ЛПНП), значительно меньше – липопротеиды промежуточной плотности (ЛППП), еще меньше – липопротеиды высокой плотности (ЛПВП2 и ЛПВП3), менее всего – липопротеиды очень низкой плотности (ЛПОНП) и хиломикроны (данные Таблицы 4).
Синтезируемый печенью холестерин поступает в кровь в составе ЛПОНП, где под влиянием липопротеинлипазы расщепляется до ЛППП. ЛППП захватываются печеночными клетками и периферическими тканями, включая макрофаги. Поступление ЛППП и ЛПНП в печеночную клетку – рецептор-опосредованный процесс. Количество рецепторов в печеночной клетке в значительной мере генетически детерминировано, и при их недостатке возникает один из вариантов наследственной гиперхолестеринемии. В печени из ЛПНП образуются ЛПВП. Значительная часть ЛПНП и ЛППП метаболизируется до желчных кислот.
Таблица 4
Содержание холестерина и триглицеридов в крови
| Показатели | Норма | Пограничный уровень | Повышенный уровень |
| Холестерин | <200 мг% | 200-239 мг% | ≥240 мг% |
| <5,2 ммоль/л | 5,2-6,2 ммоль/л | >6,2 ммоль/л | |
| Триглицериды | <200 мг% | 200-400 мг% | >400 мг% |
| <2,26 ммоль/л | 2,26-4,52 ммоль/л | >4,52 ммоль/л |
Из всех ЛП наиболее атерогенными являются ЛПНП. В то же время ЛПВП обладают антиатерогенным действием, поскольку при поступлении в клетку (макрофаг) они способны захватывать холестерин и выводить его из клетки. Среди двух фракций ЛПВП наиболее активным антиатерогенным действием обладает фракция ЛПВП3, частицы которой по размеру меньше таковых фракции ЛПВП2 (Таблица 5).
Вскоре была создана экспериментальная модель атеросклероза, которую удалось воспроизвести у кроликов путём продолжительного скармливания им холестерина. Авторами этой модели стали С.С. Халатов и Н.Н. Аничков, выдвинувшие в 1912 году инфильтрационную теорию атерогенеза. Согласно оригинальной трактовке инфильтрационный механизм состоит в том, что плазменные липиды, содержащие холестерин, просачиваются по направлению от эндотелия к адвентиции. В норме они используются для энергетических целей или удаляются через лимфатическую систему. При избытке поступления липидов и, особенно, холестерина, механизмы удаления не поспевают за накоплением, и формируется липидоз – первая стадия атеросклероза. «Без холестерина не может быть атеросклероза», – писал Н.Н. Аничков, подчеркивая решающую роль данного фактора риска как наиглавнейшего.
Таблица 5
Липидный состав липопротеидов плазмы
| Тип | Тригли- | Холестерин | Фосфолипиды | Процент |
| цериды | липопротеидов | холестерина | эфиров | |
| ЛПОНП | ||||
| ЛППП | ||||
| ЛПНП | ||||
| ЛПВП2 | ||||
| ЛПВП3 | ||||
| Хиломикроны |
На сегодняшний день разных экспериментальных моделей атеросклероза известно, но крайней мере, восемь. Это, само по себе, свидетельствует о полиэтиологической природе этого заболевания. Однако, роль дислипопротеинемий и холестерина в происхождении атеросклероза столь велика, что и сейчас в современную эпоху продолжает господствовать все тот же канонический, правда, уточненный принцип Н.Н. Аничкова: «Без атерогенных липопротеидов не будет атеросклероза».
Среди других известных экспериментальных моделей атеросклероза назовем следующие:
1. Модель Стайнера-Кендалла – первая алиментарная холестериновая модель на плотоядных животных – собаках. В течение долгого времени не удавалось получить алиментарный холестериновый атеросклероз у плотоядных животных. Причина неудач не была случайной – плотоядные обладают высоким уровнем дренажных антиатерогенных ЛПВП в крови. Стайнер и Кендалл решили эту задачу, параллельно вызвав гипотиреоз у собак метилтиоурацилом.
2. Модель Вигланда-Мальмроса также алиментарная. Однако в диету травоядных (кроликов) вводился не холестерин, а насыщенные триглицериды. Этой моделью было доказано, что холестерин в атероматозных отложениях может быть эндогенным, полученным из экзогенных предшественников. Противопоставления модели Вигланда-Мальмроса модели Халатова-Аничкова оказались несостоятельными, так как было доказано, что при избыточном введении в организм любого компонента ядра липопротеидов усиливается синтез всех липидов, необходимых для формирования их частиц, а, значит, в обеих моделях развиваются триглицеридемия и гиперхолестеринемия.
3. Существует и стрессорная (нейрогенная) теория атеросклероза. Дело в том, что теоретические взгляды авторов прототипической и алиментарной модели атеросклероза Аничкова-Халатова разошлись. Если первый придавал большое значение экзогенной холестериновой нагрузке и инфильтративному механизму, то второй считал, что атеросклероз возникает и в результате нейроэндокринных механизмов обмена эндогенного холестерина, модифицировав инфильтративную теорию в инфильтративно-комбинационную.
Первые доказательства правоты С.С. Халатова были получены Н.Т. Шутовой и П.В. Горизонтовым (1940), которые продемонстрировали, что различные воздействия на ЦНС вызывают глубокие нарушения холестеринового обмена, даже без избытка этого стероида в диете. В.П. Горизонтов и С.С. Халатов выдвинули гипотезу о роли ЦНС как депо, снабжающего избытком холестерина кровь при нервном напряжении. В годы Великой Отечественной войны Ф. Благе показал, что у умерших узников нацистского лагеря Дахау, подвергавшихся тяжелейшим хроническим стрессам, выраженный атеросклероз присутствовал даже, несмотря на резкое обеднение их тюремной диеты холестерином и насыщенными жирами. Позже, развивая идеи С.С. Халатова, была создана оригинальная модель воспроизведения атеросклероза у травоядных и плотоядных животных с помощью хронического стресса (по терминологии того периода, «нервного напряжения и перенапряжения»), или путём комбинации нейрогенного фактора и эндокринных нарушений (гипотиреоза, либо кастрации) без алиментарной липидной нагрузки. Стрессорной по своему содержанию была и модель атеросклероза, полученная у кроликов путём продолжительной иммобилизации животных.
4. Генетическая модель атеросклероза, по Ватанабэ, была воспроизведена на чистой линии гомозиготных кроликов с дефектом рецептора апо-В/Е-зависимых липопротеидов, аналогичным наблюдаемому у пациентов с семейной наследственной гиперхолестеринемией (ГЛП IIa). У таких животных безо всякой липидной нагрузки уровень холестерина в крови оказался повышен в 6-13 раз, по сравнению с нормальным. Тяжелый атеросклероз, очень похожий по морфологии на человеческий, развивался уже на протяжении 2-3 месяцах жизни, а через 5 месяцев регистрировалась ИБС. Животные гибли от инфаркта миокарда, не доживая до трехлетнего возраста. Модель Ватанабе явилась ярким подтверждением справедливости представлений М. Брауна и Дж. Гольдштейна о взаимодействии клеток сосудистой стенки и атерогенных липопротеидов.
5. Атеросклероз получен у трансгенных мышей, которым пересажен ген аномального апопротеина Е, что еще раз доказывает роль наследственных дислипопротеинемий в генезе данного заболевания.
Ряд авторов разработал модели атеросклероза, основанные на парентеральном введении атерогенных липопротеидов. Эти модели важны как свидетельство определенной роли иммунологических процессов и повреждения липопротеидных частиц в развитии атеросклероза.
6. Отечественные исследователи показали возможность воспроизведения атеросклеротических поражений сосудов у кроликов путем иммунизации их липопротеидами. Оказалось, что атеросклероз возникает и при ежедневном парентеральном введении в течение 6 месяцев липопротеидов (ЛПНП и ЛПОНП) больных кроликов, получавших пищевую холестериновую нагрузку, здоровым животным.
7. Существует модель атеросклероза X. Баумгартнера, основанная на ускорении атеросклеротического процесса в сосуде, деэндотелизированном с помощью катетера с надувным резиновым баллоном. Автор считал основным фактором увеличение инфильтрации сосуда липопротеидами при снятии эндотелиального барьера. Впоследствии было установлено, что в ходе таких манипуляций развивается артериит, и освобождаются цитокины, в том числе тромбоцитарные факторы роста, способствующие пролиферативно-склеротическому ответу сосудистой стенки, а максимальное накопление липопротеидов идет как раз не пассивным путем при отсутствующем эндотелии, а в фазу регенерации эндотелиоцитов. Важно отметить, что развитие атеросклероза по данной модели тормозится при введении животному антитромбоцитарной сыворотки.
8. Некоторые другие модели атеросклероза, оттеняющие роль тромбогенных факторов и моноклональных процессов в его развитии представлены в разделе «Патогенез атеросклероза».
Основным результатом моделирования атеросклероза и множества эпидемиологических исследований было выделение факторов риска этой болезни.
В настоящее время установлена связь атеросклероза с 246 различными факторами риска. Некоторые из них практически не подлежат профилактической коррекции или отмене, например, принадлежность к мужскому полу, наличие ряда неизлечимых генетических заболеваний (некоторых дислипопротеинемий, гомоцистинурии, порфирии). Ничего не поделаешь и с такой отчетливой тенденцией, как учащение и отягощение атеросклероза с возрастом. Однако многие важные факторы риска поддаются превентивным воздействиям, поэтому для врача важно быть информированным об этиологии и ранних признаках атеросклероза. Главными среди них следует считать (5)
1. Дислипопротеинемии (как наследственные, так и приобретенные),
2. Гипертензия (особенно, у лиц старше 50 лет),
3. Курение (притом, в первую очередь, сигарет),
4. Сахарный диабет, особенно, инсулинзависимый тип,
5. Принадлежность к мужскому полу.
В списке основных мягких факторов риска значатся ожирение (особенно абдоминального типа), гиподинамия, хронический стресс и соревновательно-стрессорный тип жизнедеятельности, по Фридману, гиперурикемия, переедание сладкого, гиперинсулинизм, гипергомоцистеинемия и фолациновый гиповитаминоз, гипервитаминоз D, использование пероральных противозачаточных средств, мягкая вода, геохимические особенности, связанные с обеспеченностью организма рядом микроэлементов, тромбофилитические состояния и т.д.
Дислипопротеинемии. Главными среди них являются:
1. Высокое содержание апопротеина (а).
2. Генетические аномалии других апопротеинов, в частности, апопротеина Е при гиперлипопротеинемии (ГЛП III), апопротеинов, кодируемых 11-ой хромосомой при гипоальфа-липопротеинемиях.
3. Высокий уровень ЛПНП (при ГЛП IIа и IIb типа), ЛППП (при ГЛП III типа), ЛПОНП (при ГЛП IIb типа, а также, в меньшей степени, ГЛП IV и V типа) и, наконец, остаточных частиц ХМ (при ГЛП III типа).
4. Понижение уровня ЛПВП.
ГЛП с повышением общего и свободного холес