Этиология
Причиной заболевания является, чаще всего, нонсенс-мутация (возникновение стоп-кодона) в гене TCOF1, приводящая к гаплонедостаточности. Ген TCOF1 расположен в локусе 5q32-33, на длинном (q) плече 5-й хромосомы, начинается от 149 737 202-й пары оснований и заканчивается на 149 779 871-й паре оснований. Продукт гена TCOF1 — ядерный транспортный белок, который экспрессируется во многих тканях во время эмбрионального и постэмбрионального развития и принимает участие в транскрипции ДНК. При синдроме Тричера Коллинза развивается состояние, при котором половинного количества генного продукта недостаточно для нормального функционирования организма.
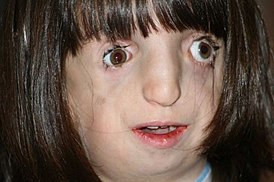
Синдром наследуется по аутосомно-доминантному принципу и характеризуется высокой пенетрантностью. Экспрессивность может быть от умеренной до выраженной, потому тяжесть дефекта отлична у разных пациентов — от почти незаметных признаков до крайне тяжёлых форм. У большинства пациентов слаборазвитые лицевые кости, что приводит к «затонувшему» лицу, крупный нос и очень маленькие челюсти и подбородок (микрогнатия). У некоторых больных присутствует волчья пасть. В тяжелых случаях микрогнатия может вытеснять язык пострадавших новорожденных достаточно, чтобы вызвать преграду ротоглотки и потенциально опасных для жизни заболеваний дыхательных путей. Врождённый порок сердца является необычной особенностью.
Характерные симптомы
У людей с СТК отмечается характерный лицевой дизморфизм с двусторонней симметричной гипоплазией скуловых костей, характерна гипоплазия инфраорбитального края глазницы с формированием антимонголоидного разреза глаз и гипоплазией нижней челюсти, что приводит к аномалии прикуса, также наблюдается апертогнатия (так называемый открытый прикус). Описана атрезия хоан, колобома (расщелина) нижних век между внешней и средней третью, сопровождающаяся отсутствием ресниц. Гипоплазия мягких тканей преимущественно отмечается в скуловой области, нижнем орбитальном крае и щеках. К особенностям относятся сложные нарушения в строении височно-нижнечелюстного сустава, что приводит к ограниченной возможности открытия рта различной степени тяжести.
|
|
Часто отмечается аномалия наружного уха, например микротия или анотия, атрезия наружного слухового прохода и аномалии развития слуховых костей, что приводит к кондуктивной тугоухости. Снижение зрения, вплоть до полной его потери. Нёбо высокое, имеет готическую форму и иногда наблюдается его расщелина.
Умственные способности, как правило, нормальные. Умственная отсталость встречается лишь у 5% людей с СТК. Из-за узких верхних дыхательных путей и ограниченного открывания рта в раннем возрасте могут возникать трудности с дыханием и питанием. Из частых признаков описан чрезмерный рост волос на щеках.


Диагностика
1. Осмотр, сбор анамнеза. Определяются характерные черепно-лицевые аномалии: недоразвитость костей скул и челюсти, деформация и гипоплазия ушных раковин, антимонголоидный тип глазных щелей, нарушения слуха и дефект верхнего неба. Иногда подтвержденный диагноз синдрома имеется у одного из родителей.
2. Биогенетический тест. Антенатальное исследование включает молекулярный анализ образца ворсин хориона на 10-11 неделе беременности, фетоскопию и анализ крови из сосудов плаценты на 18-20 неделе. После родов выполняется забор крови из вены ребенка. В обоих случаях исследуется ген TCOF1. Заболевание подтверждается при наличии в нем мутации любого типа.
|
|
3. Лучевые методы.
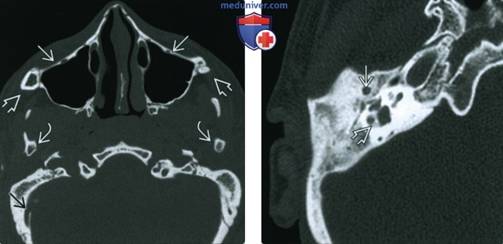 Слева: при аксиальной КТ в костном окне у девочки 13 лет с СТК определяется гипоплазия скулового комплекса с отклонением верхней челюсти кзади, отсутствием скуловых дуг, гипоплазией мыщелков нижней челюсти. Атрезия НСК, отсутствие пневматизации сосцевидного отростка, увеличение эмиссарной вены сосцевидного отростка.
Слева: при аксиальной КТ в костном окне у девочки 13 лет с СТК определяется гипоплазия скулового комплекса с отклонением верхней челюсти кзади, отсутствием скуловых дуг, гипоплазией мыщелков нижней челюсти. Атрезия НСК, отсутствие пневматизации сосцевидного отростка, увеличение эмиссарной вены сосцевидного отростка.
Справа: при аксиальной КТ в костном окне у девушки 16 лет с СТК определяется атрезия НСК, отсутствие пространства среднего уха и слуховых косточек, вентральное смещение нисходящего канала лицевого нерва, мальформация латерального полукружного канала с маленькими костными островками
4. Дородовое УЗИ. С 20-24 недели беременности ультразвуковое исследование плода способно выявить типичные изменения лица. Наиболее четко заметна двусторонняя аномалия ушей, гипоплазия скул и челюсти.
Пренатальная диагностика СТК
Несмотря на давно описанный в литературе и хорошо известный врачам-генетикам диагноз, количество статей, посвященных случаям дородовой диагностики СТК, весьма ограничено. Это связано с трудностью визуализации и объективизации некоторых классических фенотипических признаков синдрома при проведении пренатальной эхографии. Ультразвуковые проявления изменений лицевого фенотипа у плодов бывают не очевидны, и часто рождение таких детей является полной неожиданностью не только для их родителей, но и для врачей пренатальной диагностики. Явные после рождения «ядерные» признаки СТК, такие как гипоплазия скуловых костей, микрогнатия, расщелина нёба, колобома нижнего века, антимонголоидный разрез глаз, отсутствие ресниц, чаще всего остаются незамеченными, даже при современных возможностях ультразвуковых приборов, особенно когда нет генетической настороженности при осмотре, что бывает при возникновении мутации de novo у фенотипически здоровых родителей. Часто в пренатальном периоде могут наблюдаться многоводие и задержка роста плода. Внедрение в клиническую практику современных режимов сканирования при помощи объемной визуализации лицевого фенотипа значимо облегчает диагностику. Положение глазных щелей, аномальная форма носа, низко расположенные уши – все эти хорошо известные основные признаки СТК очень сложно уверенно визуализировать в обычном рутинном 2D-режиме, но при применении 3D-технологий их дефиниция становится более очевидной.
|
|
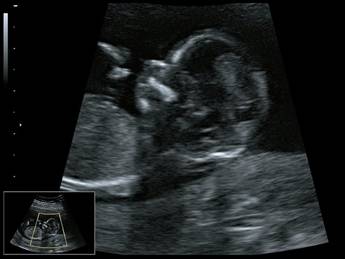
Микрогнатия - саггитальный скан в 2D, беременность 13 недель
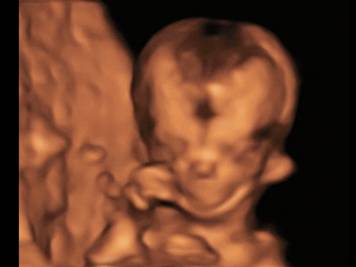
Микрогнатия - 3Dмодель лица плода
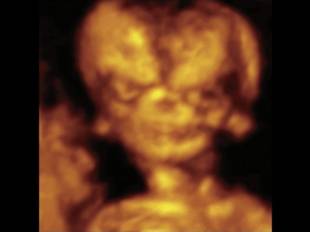
Треугольная форма лица при синдроме Тричера-Коллинза, 3Dмодель
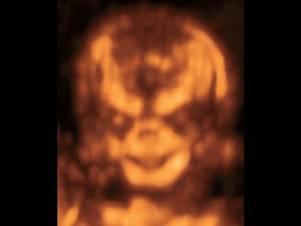
Опущенные книзу глазницы, гипоплазия скуловых костей