РОССИЙСКОЙ ФЕДЕРАЦИИ
Федеральное государственное бюджетное образовательное учреждение высшего образования
«Чувашский государственный университет имени И.Н. Ульянова»
Химико-фармацевтический факультет
Кафедра органической и фармацевтической химии
РАСЧЕТНО-ГРАФИЧЕСКАЯ РАБОТА
на тему:
«Синтез антипирин»
Выполнил: студент группы
ЗХ-42-17 Бычкова Т.В.
Проверил: ассистент
Григорьев А. А.
Чебоксары – 2020 г.
ВВЕДЕНИЕ
Производные пиразолина относятся к старейшим противовоспалительным и обезболивающим лекарственным веществам. Они синтезированы на основе включения в пиразолиновый цикл ацетанилидной фармакофорной группы, проявляющей болеутоляющее и жаропонижающее действие.
К ним относятся амидопирин (пирамидон), анальгин, бутадион. Их используют при лечении опасных гипертермии.
В последние годы в результате серьезных исследований обнаружена возможность канцерогенного влияния этих препаратов. В экспериментах на животных обнаружена возможность канцерогенного влияния амидопирина при длительном применении, а также его повреждающее действие на кроветворную систему. Фенацетин может оказывать нефротоксическое действие. В связи с этим применение этих препаратов стало ограниченным, а ряд готовых лекарственных средств, содержащих эти препараты, исключен из номенклатуры лекарственных средств (растворы и гранулы амидопирина, амидопирин с фенацетином и т. д.). Использутся до настоящего времени новомигрофен, амидопирин с бутадионом и др.
Целью настоящей работы является рассмотрение лекарственных препаратов производных пиразола на примере антипирина, подробное изучение схемы и этапов его синтеза.
Общие характеристики препарата антипирина
Антипирин является одним ин производных пиразолона. Препараты этой группы оказывают болеутоляющее, жаропонижающее и противовоспалительное действие. По анальгезирующей и жаропонижающей активности они близки к производным салициловой кислоты.
Антипирин – кристаллы слабогорького вкуса, без запаха, хорошо растворим в воде (1:1), в спирте (1:1), в хлороформе (1:15) хуже в эфире (1:75). Дает все характерные качественные реакции на алкалоиды. С FeCl3 дает интенсивное красное окрашивание. Качественной реакцией на антипирин является изумрудная окраска нитрозоантипирина.
Антипирин обладает умеренным противовоспалительным действием; более активны амидопирин, анальгин и особенно бутадион. При местном применении антипирин оказывает также некоторое кровоостанавливающее действие. Применяют антипирин внутрь при невралгиях, ревматизме, хорее, простудных заболеваниях: Назначают взрослым по 0,25-0,5 г; детям старше 6 месяцев — по 0,03-0,25 т на прием в зависимости от возраста. Принимают 2-3 раза в день. Высшие дозы для взрослых внутрь: разовая 1 с, суточная 3 г. Как кровоостанавливающее средство антипирин иногда применяют (10—20% раствор) для смачивания тампонов и салфеток при носовых и паренхиматозных кровотечениях. При назначении антипирина следует учитывать возможность повышенной чувствительности больных к препарату с появлением крапивницы и фиксированной сыпи.
Формы выпуска: порошок и таблетки по 0,25 г. Хранение: список Б. В хорошо укупоренной таре, предохраняющей от действия света.
Получение антипирина
Широкое изучение пирозолоновых соединений и открытие их ценного фармакологического действия связаны с синтетическими исследованиями в области хинина.
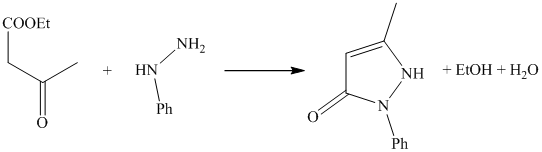
Стремясь получить тетрагидрохинолиновые соединения, обладающие жаропонижающими свойствами хинина Кнорр в 1883 г. осуществил конденсацию ацетоуксусного эфира с фенилгидрозином, который проявляет слабое жаропонижающее действие, плохо растворим в воде; метилирование его привело к получению высокоактивного и хорошо растворимого препарата 1-фенил-2,3-диметилпирозолона (антипирина).
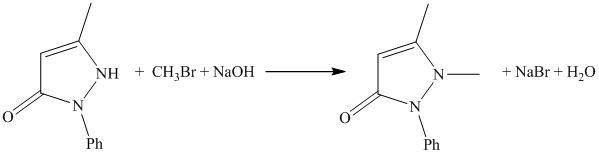
Современное промышленное производство антипирина осуществляется из более дешевого сырья — дикетена, который является продуктом пиролиза ацетона (при 500—600 °С над окисью алюминия). Дикетен конденсируют с фенилгидразином. Образовавшийся 1-фенил-3-метилпиразолон-5 метилируют, используя в качестве метилирующего агента метиловый эфир бензолсульфокислоты, который дает возможность увеличить выход антипирина до 90%, не используя при этом высокое давление. Антипирин не только сам является лекарственным препаратом, но и служит исходным продуктом для получения амидопирина и анальгина.
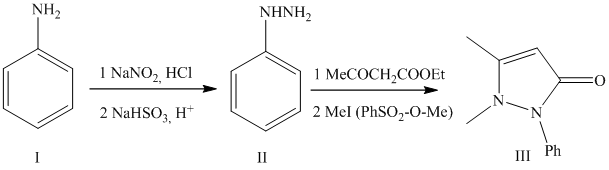
В эмалированный реактор с масляным обогревом загружают фенилметилпиразолон и проводят его сушку в вакууме при 100оС до полного удаления влаги. Затем повышают температуру до 127-130оС и к раствору фенилметилпиразолона приливают метиловый эфир бензосульфокислоты. Температура реакции не выше 135-140оС. По окончании процесса реакционную массу передавливают в кристаллизатор, куда загружают небольшое количество воды и охлаждают до 10оС. Выпавший бензосульфонат антипирина отжимают и промывают на центрифуге. Для выделения антипирина, эту соль обрабатывают водным раствором NaOH, полученный антипирин отделяют от раствора соли переосаждают в изопропиловом спирте, антипирин очищают перекристаллизацией из изопропилового спирта. Выпускается в порошках и таблетках по 0,25 г.
Возможные побочные процессы в синтезе антипирина
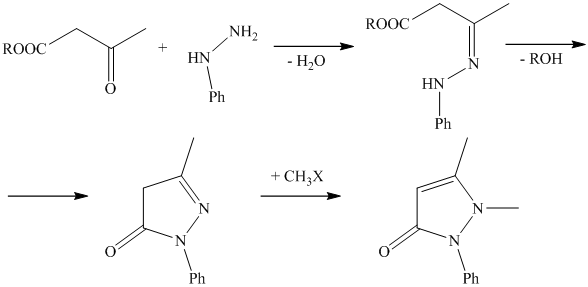
Учитывая наличие кето-енольной таутомерии ацетоуксусного эфира, а также таутомерию в пиразолоновом ядре, при рассмотрении реакции между фенилгидразином и ацетоуксусным эфиром можно предположить образование нескольких изомерных форм 1-фенил-3-метилпиразолона.
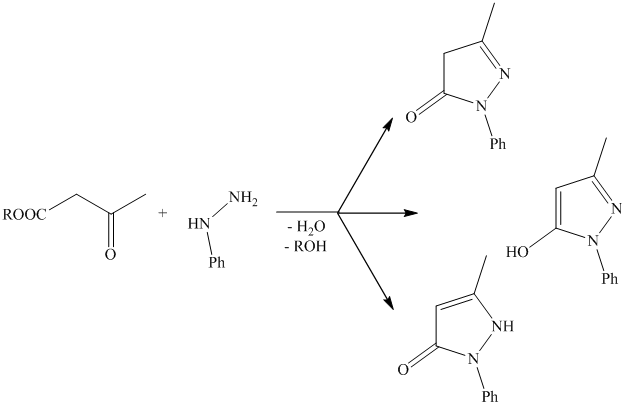
Однако, 1-фенил-3-метилпиразолон известен лишь в 1 форме. Бесцветные кристаллы, температура поавления – 127оС, температура кипения – 191оС.
Процесс метилирования фенилметилпиразолона может быть представлен через промежуточное образование четвертичной соли, которая под действием щелочи перегруппировывается в антипирин.
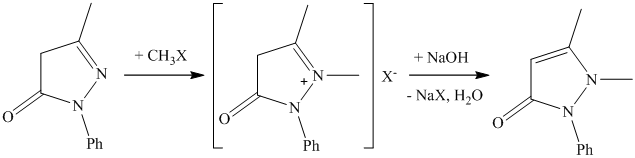
Строение антипирина подтверждено встречным синтезом при конденсации енольной формы ацетоуксусного эфира или галоидного эфира с метилфенилгидразином, так как положение обеих метильных групп определяются исходными продуктами.
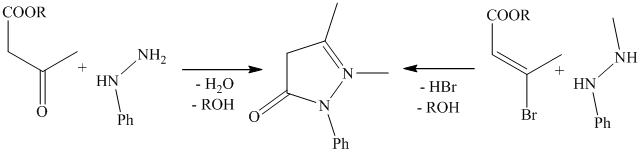
Как способ получения не применяется, так как низкий выход и малодоступные продукты синтеза.
Реакцию ведут в нейтральной среде. Если осуществить реакцию в кислой среде, то при температуре происходит отщепление не спирта, а второй молекулы воды, и образуется 1-фенил-3-метил-5-этоксипиразол.
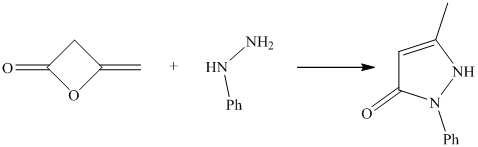
Для получения 1-фенил-3-метилпиразолона являющегося важнейшим полупродуктом в синтезах пиразолоновых препаратов разработан также метод, в котором используется дикетон.
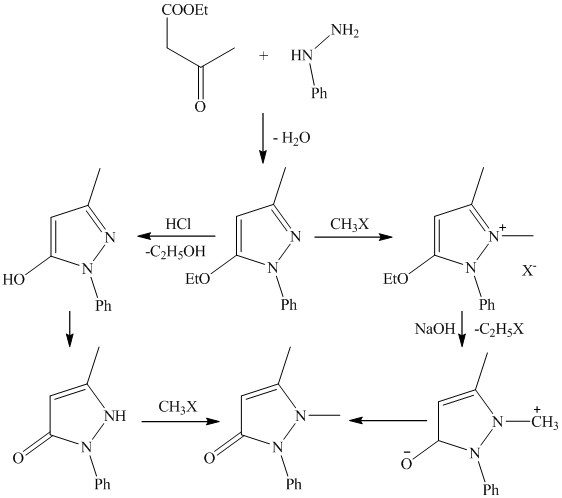
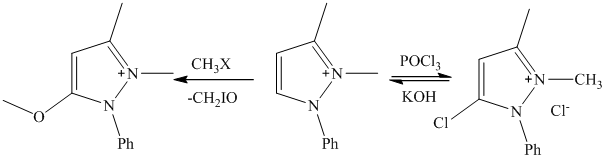
Свойства антипирина – высокая растворимость в воде, реакции с иодистым метилом, РОСl3 и др. объсняется тем, что он имеет структуру внутреннего четвертичного основания.
В промышленном синтезе антипирина помимо важного значения условий проведения основной конденсации между ацетоуксусным эфиром и фенилгидразином (выбор среды, нейтральная реакция, небольшой избыток фенилгидразина и т. д.) определенную роль играет выбор метилирующего средства.:
Диазометан не пригоден, так как приводит к о-метиловому эфиру четвертичной соли, частично образующимся и при метилировании иодистым метилом.
Лучше для этих целей использовать хлористый или бромистый метил, диметилсульфат или, лучше, метиловый эфир бензолсульфокислоты, так как в этом случае нет необходимости в поглощающих автоклавах (СН3Вr – 18 атм.; СН3Сl – 65 атм.).
Очистка получаемого антипирина обычно осуществляется 2-3 кратной перекристаллизацией из воды; может быть использована перегонка в вакууме (200-205 оС при 4-5 мм, 141-142 оС в вакууме катодного свечения).
Стадии синтеза антипирина
Стадии синтеза
1. Фенилгидразин.
2. Ацетоуксусный эфир.
3. Фенилметилпиразолон.
4. Фенилдиметилпиразолон (антипирин).
1 стадия
В стеклянную банку емкостью в 3 л, снабженную механической мешалкой и капельной воронкой, вносят 93 г свежеперегнанного анилина, 286 г технической соляной кислоты и 90 г льда. Реакционную смесь охлаждают смесью льда и поваренной соли до 0-5° и медленно при хорошем перемешивании приливают из капельной воронки водный раствор нитрита натрия (72 г нитрита в 110 см3 воды). Температуру реакционной массы поддерживают на нуле и во всяком случае не выше 3-5°. По мере хода реакции прибавляют еще около 100 г льда. О конце реакции судят по посинению иод-крахмальной бумажки.
Пока идет процесс диазотирования, а еще лучше до начала его, заготовляют водный раствор сернистокислого натрия.
В колбе емкостью в 3 л растворяют 280 г технического едкого натра в 640 см 3 воды и в полученный раствор пропускают при охлаждении ток сернистого ангидрида до появления нейтральной реакции на фенолфталеин, причем нейтральность устанавливают в отдельной пробе, предварительно разбавленной водой; окраска должна быть слаборозовая.
После установления нейтральности пропускают сернистый ангидрид в течение еще 2-3 мин.
Полученный раствор сернистокислого натрия охлаждают до -2° и к нему приливают при хорошем перемешивании возможно быстрее раствор хлористого диазобензола. В реакционную смесь во время хода реакции прибавляют 60 г льда. Выпадает сернистокислая соль диазобензола в виде объемистого желтого осадка. Реакционную массу начинают медленно нагревать, доводят до 60° и эту температуру поддерживают в течение 1 часа. После этого пропускают в жидкость при той же температуре ток сернистого ангидрида до полного растворения осадка и оставляют стоять на несколько часов. Полученный таким образом раствор сульфоната фенилгидразина фильтруют, нагревают до 50-55° и при помешивании прибавляют к нему 600 г технической соляной кислоты. Снова нагревают в течение получаса и затем охлаждают до 0°. Выпадают бледнорозовыё кристаллы солянокислого фенилгидразина. Их отсасывают на бюхнеровской воронке.
Полученную соль фенилгидразина помещают в колбу емкостью в 1 л, прибавляют 375 г 25%-ного раствора едкого натра или раствора кристаллической соды (270 г соды на 400 см 3 воды); выделяется светложелтое маслооснование фенилгидразина. Его отделяют в делительной воронке от слоя воды; водный слой экстрагируют дважды бензолом и вытяжку соединяют с основной массой фенилгидразина, отделенной от воды.
Бензольный раствор сушат твердым едким натром. Отгоняют бензол на водяной бане при обыкновенном давлении, а фенилгидразин перегоняют в вакууме при 10 мм давления.
Температура кипения при 10 мм 116-118°, при 18 мм 137-138°.
При охлаждении фенилгидразин закристаллизовывается. Температура плавления кристаллического фенилгидразина 23°.
Выход 77 г, что составляет 71,5-72,5% теории.
2 стадия
В колбу емкостью в 1 л, снабженную мешалкой, обратным хо, лодильником и вводной трубкой для СО2, загружают 130 г ксилола- предварительно высушенного над хлористым кальцием и перегнанного, и 26 г металлического натрия. Колбу нагревают до 80-90°, причем металлический натрий расплавляется; понизив температуру до 50°, пускают мешалку; натрий раздробляется в пыль. С раздробленного натрия сливают почти весь ксилол, вводят СО2 и вливают через холодильник уксусный эфир. Уксусноэтиловый эфир предварительно нейтрализуют содой, промывают водой, сушат в течение 2 суток над хлористым кальцием и перегоняют. После введения эфира реакционную смесь слегка нагревают. Металлический натрий успевает прореагитовать приблизительно в течение 1 часа. Затем реакционную массу охлаждают до 20° и вводят смесь уксусной кислоты с водой (52 г кислоты и 140 г воды,; прибавляют равный обьем насыщенного на холоду раствора поваренной соли и разделяют образовавшиеся два слоя в делительной воронке. Верхний слой, состоящий из ацетоуксусного эфира и уксусного эфира, промывают небольшим количеством воды и отгоняют уксусноэтиловый эфир при нормальном давлении (до 80-90°), а затем остаток перегоняют в вакууме.
Выход ацетоуксусного эфира 92,6 г, т. е. 32% теории.
3 стадия
В колбу емкостью в 350 см3, снабженную обратным холодильником, мешалкой и капельной воронкой, загружают 77 г свежеперегнанного фенилгидразина и при хорошем перемешивании добавляют из капельной воронки 92,6 г свежеперегнанного ацетоуксусного эфира, предварительно разбавленного 10 г 96%-ного этилового спирта.
Реакционною смесь нагревают на водяной бане, пока взятая проба не будет целиком застывать в кристаллическую массу, что достигается после нагревания приблизительно в течение 4-6 час. Далее, содержимое колбы выливают в фарфоровую чашку; при охлаждении полученный фенилметилпиразолон закристаллизовывается. Кристаллическую массу отсасывают на бюхнеровской воронке и промывают 96%-ным этиловым спиртом до белого цвета. Если фенилметилпиразолон остается окрашенным, его перекристаллизовывают из спирта же, причем на каждые 1 вес. ч. продукта берут 6 вес. ч. спирта.
Из маточников отгоняют спирт и полученное дополнительнее количество фенилметилпиразолона снова перекристаллизовывают.
Белые кристаллы, планятся при 127°.
Выход 87 г, что соответствует 90% от теории.
4 стадия
Метилирование фенилметилпиразолона производят или а) метиловым эфиром паратолуолсульфокислоты или б) бромистым метилом.
а) Метилирование метиловым эфиром паратолуолсульфокислоты
Стадия метилирования фенилметилпиразолона состоит из следующих двух фаз:
1) Получение метилового эфира паратолуолсульфоновой кислоты.
2) Действие метилового эфира паратолуолсульфоновой кислоты на фенилметилпиразолон.
1) Получение метилового эфира паратолуолсульфоновой кислоты
В колбу или стакан емкостью в 1 л, снабженные мешалкой и капельной воронкой, загружают 118 г предварительно отмытого и высушенного паратолуолсульфохлоридаР к толуолсульфохлориду приливают 65 г метилового спирта и при хорошем помешивании и охлаждении до 5 — 7° приливают из капельной воронки 25%-ный раствор едкого натра (105 г). После прибавления всей щелочи смесь продолжают размешивать в течение 2-3 час. Далее реакционную массу охлаждают да 0°. Спустя несколько часов метиловый эфир паратолуолсульфокислоты выпадает в виде мелких кристаллов. Их отсасывают на бюхнеровской воронке, нагревают до расплавления и фильтруют. На фильтре остается поваренная соль и непрореагировавшие кристаллы толуолсульфохлорида.
Для получения эфира в кристаллическом виде фильтрат снова замораживают.
Выход 93-95 г, что соответствует 80-85% теории
2) Метилирование фенилметилпиразолона
В круглодонную колбу емкостью в 350 см3, снабженную мешалкой и холодильником, загружают 87 г фенилметилпиразолона и 93 г метилового эфира паратолуолсульфокислоты и нагревают на масляной бане при хорошем размешивании до 135-140° в течение 40-45 час. Затем реакционную массу охлаждают до 95-100° и при этой температуре прибавляют около 100 см3 воды и снова перемешивают в течение получаса, после чего выливают в фарфоровую чашку; паратолуолсульфонат фенилметилпиразолона на холоду закристаллизовывается. Его отсасывают на бюхнеровской воронке, промывают дважды водой и сушат при 40-50°.
К полученному сульфонату фенилметилпиразолона приливают 100 г воды и постепенно вносят раствор кристаллической соды (70 г соды в 100 см3 воды). Полученный раствор антипирина и натриевой соли паратолуолсульфоновой кислоты фильтруют и далее многократно экстрагируют бензолом (бензола расходуется приблизительно 1—1% кг). Вытяжки соединяют вместе и почти полностью отгоняют растворитель до получения весьма концентрированного раствора антипирина, который по охлаждении закристаллизовываетея в виде мелких, слегка окрашенных кристаллов. Их перекристаллизовывают из воды (на каждую весовую часть антипирина берут 0,5 ч. воды), к которой добавляют небольшое количество бисульфита (несколько кубических сантиметров) и животного угля. Полученные кристаллы отфильтровывают, а фильтрат упаривают. Антипирин, полученный из маточника, перекристаллизовывают 2-3 раза.
Выход 75 г, что составляет 80% теории.
Антипирин может быть перекристаллизован также из спирта причем на 1 вес. ч. антипирина берут 0,3 вес. ч. спирта. Кристаллизацию ведут медленно.
б) Метилирование бромистым метилом
Реакцию ведут в эмалированном автоклаве емкостью в 1 л, в который загружают 87 г фенилметилпиразолона, 30 г метилового спирта и 48 г бромистого метила и нагревают до 118-130° в течение 17 час, причем давление доходит до 2,5 ат. Затем автоклав охлаждают, осторожно выпускают образовавшийся НВг, который поглощают в промывалках со щелочью. Далее реакционную массу переносят в колбу, снабженную обратным холодильником и мешалкой, и нагревают около 3 час. при температуре 115°. После нагревания переставляют обратный холодильник на прямой и отгоняют спирт. По окончании перегонки реакционную массу охлаждают примерно до 50°, прибавляют щелочь до щелочной реакции (около 50 г 40°/0-ного едкого натра) и около 500-1000 г бензола и снова нагревают до 50° в течение 5 час.
Образовавшиеся два слоя разделяют в делительной воронке. Нижний водный слой снова экстрагируют бензолом (500 г). Бензольные вытяжки соединяют и отгоняют бензол. Полученный антипирин перекристаллизовывают из воды или спирта.
Белый кристаллический порошок; темп. пл. 113°, темп. кип. 211-212° при 10 мм.
Выход около 84 г, что соответствует 90% теории.
РАСЧЕТЫ
Для синтеза антипирина нами было взято 10 кг исходного ацетоуксусного эфира.
Синтез Фенилметилпиразолона
M (ацетоуксусный эфир) = 130,14 г/моль
M (фенилгидразин) = 174,20 г/моль
m (ацетоуксусный эфир) = 10,00 кг (76,84 моль)
теоретическая m (фенилметилпиразолон) = 13,39 кг (76,84 моль)
практическая m (фенилметилпиразолон) = 12,05 кг (69,16 моль)
выход продукта = 12,05/13,39 = 0,90 или 90,0 %
Синтез Фенилдиметилпиразолон (антипирин)
M (фенилметилпиразолон) = 174,20 г/моль
M (фенилдиметилпиразолон) = 188,23 г/моль
m (фенилметилпиразолон) = 12,05 кг (69,16 моль)
теоретическая m (фенилметилпиразолон) = 13,02 кг (69,16 моль)
практическая m (фенилдиметилпиразолон) = 11,72 кг (62,24 моль)
выход продукта = 11,72/13,02 = 0,90 или 90,0 %
Суммарный выход продукта = 62,24/76,84 = 0,81 или 81,0%.
ВЫВОДЫ
1. Изучен метод синтеза антипирина на основе ацетоуксусного эфира, подробно изучены стадии синтеза и возможные побочные процессы.
2. На основе экспериментальных данных рассчитан выход антипирина, который составил 81,0%.
Список использованной литературы
1. Машковский М. Д. Лекарственные средства в 2 Т, Т.1. — 14 изд. перераб. и доп. — М. ООО «Издательство Новая волна» 540 с., с.422.
2. Клиническая фармакология: Учебник / Под ред. В. Г. Кукеса. — 3-е изд., перераб. и доп.. - М.: ГЭОТАР-Медиа, 2006. - С. 287-297. - 944 с. - ISBN 5-9704-0287-7.
3. Рубцов М.В., Байчиков А.Г. Синтетические химико-фармацевтические препараты. – М.: Медицина, 1971. –328 с.
4. Клиническая фармакология: Учебник / Под ред. В. Г. Кукеса. - 3-е изд., перераб. и доп.. - М.: ГЭОТАР-Медиа, 2006. - С. 287-297. - 944 с. - ISBN 5-9704-0287-7.
5. Яхонтов Л.Н. Синтетические лекарственные средства / Яхонтов Л.Н., Глушков Р.Г.. Под ред. Натрадзе А.Г. – М.:Медицина, 1983. 272 с.
6. Беркенгейм А.М. "Химия и технология синтетических лекарственных средств" М.:ОНТИ НКПТ, 1935.
7. Вартанян Р.С. Синтез основных лекарственных средств. — М.: МИА, 2004 - 845 с.
- Основы органической химии лекарственных веществ/ А.Т. Солдатенков, Н.М. Колядина, И.В. Шендрик - М: Химия, 2001 - 192 с.