Цель: Познакомиться на теоретическом уровне с высокоэффективной жидкостной хроматографией, и на практике, с тонкослойной хроматографией.
ВЖЭХ
Теория метода:
Хроматография – процесс разделения смеси веществ, в зависимости от сродства его к разделяющему агенту. Имеется много видов хроматографии, но все они основываются на процессах сорбции – десорбции компонентов смеси на поверхности адсорбента.
В ВЭЖХ процесс разделения протекает в трубчатых колонках длиной около 20 см, неподвижной фазой при этом служит чаще всего разного рода силикагели, либо полимеры.
Хроматограф состоит из следующих компонентов:
1. блок подачи элюентов,
- насос,
- инжектор,
- колонки,
- детектор,
- ёмкости для отработанных материалов
Элюенты представляют собой чистые жидкости, смеси жидкостей или жидкости, в которых растворены модификаторы. Для обеспечения воспроизводимости при работе состав элюента должен быть описан полно и недвусмысленно, чтобы можно было приготавливать один и тот же элюент снова и снова. Приготовленные элюенты хранят в резервуарах, откуда они поступают в насосы за счёт действия плунжера и обратного клапана. Элюент поступает из резервуара, проходит через входной фильтр и соединительный трубопровод. После того, как элюент попал в насосную систему, его называют подвижной фазой. Состав подвижной фазы может оставаться постоянным (изократическое элюирование) или изменяться (градиентное элюирование). От подвижной фазы зависит общее среднее время удерживания анализируемых веществ: при более сильном элюенте временя элюирования будет меньше, при более слабом – больше. Градиентные режимы элюирования могут быть самыми различными: линейный, экспоненциальный, ступенчатый и т.д. Столь различные градиентные режимы используются для того, чтобы как можно лучше разделить (разрешить) пики анализируемых веществ и при этом как можно более сократить продолжительность анализа.
Насос включает один или несколько узлов, состоящих из корпуса и плунжера. Диапазон скоростей подачи элюента насосом зависит от сочетания объёма полости, диаметра плунжера, длины хода плунжера и скорости его перемещения. Как правило, различают микронасосы (расход от 1 до 500 мкл/мин), аналитические (от 0,1 до 10 мл /мин) и препаративные насосы (расход свыше 5 мл/мин.
Для изократического элюирования достаточно одного насосного узла корпуса с плунжером. Для градиентного режима требуются либо несколько насосных узлов, либо ряд соленоидов, управляющих расходом элюентов из нескольких резервуаров через один насосный узел. В любом из случаев градиентный режим предполагает смешение элюентов двух или более различных составов в изменяющейся пропорции. Смешивание может осуществляться либо до того, как элюент достигает плунжера (это называется смешиванием под низким давлением), либо после плунжера (смешиванием под высоким давлением). Для смешивания под высоким давлением чрезвычайно важна дегазация, поскольку в результате подвижная фаза смешивается в малом объёме под большим давлением. Газовыделение при смешивании в этом случае вызовет серьёзные нарушения нормальной работы системы.
В современных установках для ВЭЖХ градиентный режим реализуется смешением под низким давлением в одноплунжерной системе или под высоким давлением в двухплунжерной системе. Во многих насосных системах между выходным обратным клапаном и инжектором помещают датчик давления. Этот компонент постоянно замеряет давление в системе. Непрерывный контроль давления – важный прием профилактики нарушений нормальной работы.
Проба - «матрица», содержащая вещества, подлежащие анализу, независимо от физического состояния (твёрдое, жидкое или газообразное). Проба содержит анализируемые вещества, называемые также аналитами. Кроме того, проба содержит неконтролируемые в процессе анализа вещества, которые в совокупности будут именоваться примесями. Как только проба приобретает вид, пригодный для анализа, её вводят в подвижную фазу с помощью устройства ввода пробы, также называемого инжектором. Инжектор с большой точностью отмеряет определённый объём пробы перед вводом в подвижную фазу. Проба вводится в инжектор с помощью шприца-пробоотборника.
Когда проба попадает в колонку, она взаимодействует с набивкой (сорбентом) – особым материалом содержащимся в колонке (аналогичным материалом заполнены насыщающая колонка и предколонка). Колонка в сочетании с подвижной фазой выполняет разделение анализируемых веществ.
Совместимость элюентов с колонками является, скорее, правилом, чем исключением. Однако в некоторых случаях приходится использовать агрессивные элюенты (т.е., такие, которые могут быстро вызвать необратимое повреждение). В некоторых случаях серьёзные нарушения работы могут вызывать имеющиеся в элюенте примеси. Приведём некоторые примеры подобных сочетаний элюентов и колонок.
1. Буферы, кислотные или основные, из-за которых рН элюента > 8 или < 2, при использовании с колонкой на силикагеле.
2. Элюенты с различным содержанием воды при использовании с колонкой на непривитом силикагеле или немодифицированной окиси алюминия.
3. Сополимеры полистирола и нестабилизированный тетрагидрофуран (ТГФ), содержащий пероксиды, которые вступают в химическую реакцию с полимером.
Более чем в 75% случаев в настоящее время набивкой является материал на основе пористого сферического силикагеля. Хотя в некоторых случаях силикагель используется как набивка без дополнительной обработки, в болшинстве случаев он подвергается химической модификации, чтобы получить поверхности с весьма различными полярностью и функциональными группами. Функциональные группы, привитые к поверхности, называются в совокупности неподвижной фазой. Если материал набивки не подвергался химической модификации, то неподвижная фаза и набивка – одно и то же. Однако термины «набивка» и «неподвижная фаза» не следует путать.
Именно в колонке происходит хроматографическое разделение. Хотя конструкция и внешний вид колонок разных изготовителей могут различаться достаточно сильно, многие их детали совпадают. Чаще всего детали колонки выполняют из нержавеющей стали марки 316.
Аналиты пробы имеют различное сродство к паре подвижная фаза – неподвижная фаза. Для конкретной пары подвижная/неподвижная фаза эти различия заставляет аналиты по –разному распределяться между подвижной и неподвижной фазой воспроизводимым образом. Соответственно, при движении подвижной фазы вдоль колонки, каждый аналит перемещается по колонке с разной (неповторяющейся) скоростью и выходит из колонки в определённый хорошо воспроизводимый момент времени.
Хотя общее время удерживания того или иного вещества и специфика его отделения от остальных определяется в основном составом подвижной фазы, колонка также определяет характер разделения. От размера колонки зависит также, какое количество подвижной фазы расходуется в одном анализе.
Выходящие из колонки аналиты с током элюента попадают в детектор. Детектор выбирают так, чтобы его отклик на каждый из аналитов был оптимальным. Детектор называют универсальным, если он даёт отклик на любое из проходящих по нему веществ. Таким, например, является рефрактометрический детектор. Другие детекторы реагируют на специфические свойства некоторого соединения или класса соединений. Например, спектрофотометрический детектор ультрафиолетового (УФ) диапазона реагирует на хромофоры, поглощающие свет на определённых длинах волн, электрохимический детектор измеряет силу тока, возникающую при окислении или восстановлении вещества на электроде, имеющем определённый потенциал.
На последней стадии вещество из детектора поступает в ёмкость для отработанного элюента классифицируется и утилизируется надлежащим образом.
Все перечисленные компоненты необходимы для того, чтобы правильно организовать работу и получать результаты с высокими точностью, сходимостью и воспроизводимостью.

Время удерживания, t r. Время, за которое компонент проходит всю систему от инжектора (момент ввода пробы принимается за t = 0) до детектора. Такое прохождение называется элюированием. Определяется путём опускания перпендикуляра из вершины пика на нулевую линию (рис 2).
Коэффициент ёмкости k '. Степень удерживания компонента, выраженная в кратности его времени удерживания к времени удерживания несорбируемого компонента по следующей формуле:
k' = (tr – t0) / t0.
Для пика несорбируемого компонента tr = t 0, и k' = 0. Порядок расчёта также показан на рис2.
Высота пика h p. Расстояние от вершины пика до нулевой линии. Нулевая линия определяется как обычный отклик детектора в присутствии подвижной фазы, но при отсутствии пробы. Нулевая линия может быть горизонтальной, как при изократическом элюировании, или наклонной, как в некоторых градиентных режимах. Поскольку зачастую высота пика линейно зависит от содержания компонента, измерение высоты пика может быть использовано для количественного анализа веществ.
Используются также параметры, отражающие форму пика в нескольких точках: 0,1 hp или 10% hp, 0,5 hp или 50% h p.
Ширина пика w x. Ширина пика измеряется на заданной высоте пика х. Так, ширина на половине высоты обозначается w0,5 или w50.
Число теоретических тарелок N. Число теоретических тарелок колонки является показателем эффективности разделения. По определению,
N = 5,54 (tr / tw1 /2)2,
где tr – время удерживания, tw½ – ширина пика на половине высоты.
Кроме того, N можно рассчитать по ширине пика на нулевой линии. Эта ширина измеряется между точками пересечения нулевой линии касательными, проведёнными в точках перегиба пика. Значение N при этом рассчитывается следующим образом:
N =16 (t r / t wb)2,
где tr – время удерживания, twb – ширина пика на нулевой линии.
Обратите внимание: при одном том же времени удерживания, чем меньше ширина пика (т.е., чем уже пик), тем больше число теоретических тарелок колонки. Иначе говоря, при прочих равных, чем больше N, тем эффективнее колонка.
С точки зрения контроля нормальной работы хроматографической системы, значение N крайне важно, поскольку оно зависит почти ото всех параметров системы: состава подвижной фазы (поскольку состав влияет на t r), типа, длины и внутреннего диаметра колонки (поскольку тип и размеры колонки также влияют на t r), температуры (поскольку, как правило, чем выше температура Т, тем меньше время удерживания tr и ширина пика w), объёма пробы, всех внеколоночных объёмов хроматографической установки (трубопроводы, ячейка детектора и пр.).
Степень разделения α. Данный параметр используется для характеристики разделения двух соседних пиков. В идеале эти пики не должны перекрываться, то есть должны быть разрешены до нулевой линии. Это условие выполняется для пиков сходной формы, если α >1,15. Степень разделения рассчитывается следующим образом:
α = k'2 / k'1,
где нижние индексы показывают порядок выхода компонентов (рис 2). При этом всегда α >1.

Разрешение R s. Важный параметр хроматограммы более чем двух компонентов – расстояние между любыми двумя соседними пиками. Для его оценки может, например, быть использовано разрешение R s, определяемое как
 ,
,
t1 и t2 – времена удерживания первого и второго пиков, W1= twb1 и W 2= twb2, соответственно,– ширины этих пиков, определяемые как расстояние между точками пересечения нулевой линии касательными, проведенными через точки перегиба.
Коэффициент асимметрии Ax. Коэффициент асимметрии показывает, насколько форма пика отличается от нормального распределения. Нижний индекс х указывает, на какой высоте пика определяется асимметрия. Чаще всего используются А 10 (рассчитываемая на 10% высоты пика) и А5 (на 5 % высоты пика). Коэффициент асимметрии находят из выражения
Ax = b/ a,
где b – расстояние от перпендикуляра, опущенного из вершины пика на нулевую линию, до конечной точки кривой, a – расстояние от перпендикуляра, опущенного из вершины пика на нулевую линию, до начальной точки кривой пика.
Задание:
Определить все параметры хроматограммы смеси стероидов.
t0 = 5 min
tx1 = 24,8 min
tx2 = 31,5 min
k1` = 3.96
k2` = 5.3
h1 = 40mm
h2 = 98mm
w (0.5) 1 = 1.5mm
w (0.5) 2 = 2mm
w1 = 6mm
w2 = 8mm
N = 1520
a = 1.34
Rs = 1.675
C – пропорциональна площади пика
С1 = С2*S1/S2
S1 = 120mm2
S2 = 392mm2
C2 = 100mkM
C1 = 30.6mkM
ТСХ
Теория метода
Тонкослойная хроматография (ТСХ, TLC) - один из наиболее используемых методов хроматографического анализа, но наименее популяризируемый.
Несмотря на существовавшие до недавнего времени существенные недостатки, она широко используется для качественного анализа смесей, в основном, за счет дешевизны и скорости получения результатов.
Основой тонкослойной хроматографии является адсорбционный метод, хотя также встречается метод распределительной хроматографии.
Адсорбционный метод основан на различии степени сорбции-десорбции разделяемых компонентов на неподвижной фазе. Адсорбция осуществляется за счет ван-дер-вальсовских сил, являющейся основой физической адсорбции, полимолекулярной (образование нескольких слоев адсорбата на поверхности адсорбента) и хемосорбцией (химического взаимодействия адсорбента и адсорбата).
Для эффективных процессов сорбции-десорбции необходима большая площадь, что предъявляет определенные требования к адсорбенту. При большой поверхности разделения фаз происходит быстрое установление равновесия между фазами компонентов смеси и эффективное разделение.
Так физическое выражение адсорбции-десорбции в упрощенном виде можно выразить уравнением [1]
Г=(Г~/К)с.
где Г~ -предельно возможная величина адсорбции, К- константа равновесия; с-концентрация абсорбата.
В более строгих подходах к теории адсорбции необходимо учитывать взаимодействие между адсорбированными частицами, неоднородность поверхности, давление, температуру и т.д.
Но как видно из вышеописанного уравнения адсорбция является линейной функцией концентрации.
Еще одним видом используемом в методе тонкослойной хроматографии является распределительная жидкостная хроматография.
В распределительной хроматографии обе фазы - подвижная и неподвижная - жидкости, не смешивающиеся друг с другом. Разделение веществ основано на различии в их коэффициентах распределения между этими фазами.
Тонкослойная хроматография.
В этом методе хроматографирование веществ происходит в тонком слое сорбента, нанесенного на твердую плоскую подложку. Разделение в этом методе в основном происходит на основе сорбции-десорбции.
Использование различных сорбентов, позволило значительно расширить и улучшить этот метод.
В начале появления метода пластины приходилось изготавливать самостоятельно. Но на сегодняшний день в основном используются пластины заводского изготовления, имеющие достаточно широкий ассортимент как по размерам и носителям, так и по подложкам.
Современная хроматографическая пластинка представляет собой основу из стекла, алюминия или полимера (например политерефталат). В связи с тем, что стеклянная основа становится менее популярной (часто бьется, нельзя разделить пластинку на несколько частей не повредив слой сорбента, тяжелая по весу), наибольшее распространение получили пластины, в качестве основ которых используют алюминиевую фольгу или полимеры.
Для закрепления сорбента применяют гипс, крахмал, силиказоль и др., которые удерживают зерна сорбента на подложке. Толщина слоя может быть различна (100 и более мкм), но самый важный критерий - слой должен быть равномерный по толщине в любом месте хроматографической пластинки.
Сорбенты
Наиболее распространенным сорбентом является силикагель.
Силикагель - гидратированная кремниевая кислота, образующаяся при действии минеральных кислот на силикат натрия и сушкой образовавшегося золя. После размалывания золя используют фракцию определенной зернистости (указанную на пластинке, обычно 5-20 мкм).
Силикагель является полярным сорбентом, у которого в качестве активных центров служит группы -ОН. Он легко сорбирует на поверхности воду и образует водородные связи.
Окись алюминия. Окись алюминия является слабо основным адсорбентом и используется в основном для разделения соединений слабоосновного и нейтрального характера. Недостатком пластин на окиси алюминия является обязательная активация поверхности перед использованием в сушильном шкафу при высокой температуре (100-150 0С) и низкая, по сравнению с силикагелем адсорбционная емкость слоя.
Кизельгур - адсорбент, полученный из природных минералов: диатомовых земель. Сорбент обладает гидрофильными свойствами, но более низкой адсорбционной емкостью слоя по сравнению с силикагелем.
Кремнекислый магний менее полярный чем силикагель и обычно используется в случаях, когда более полярные адсорбенты не дали эффективного разделения.
Целлюлоза - тонкослойные пластины с нанесенной целлюлозой очень эффективны для разделения сложных органических молекул. Адсорбент представляет собой в основном шарики целлюлозы диаметром до50 мкм, закрепленные на носителе крахмалом. Но как и в бумажной хроматографии, подъем фронта растворителя происходит очень медленно.
В ионообменных хроматографических пластинках в качестве адсорбента используют ионообменные смолы, содержащие четвертичный аммоний или активные сульфогруппы, участвующие в ионном обмене. Тонкослойная хроматография с такого типа пластинками, проводится с подвижными фазами содержащими сильные кислоты или щелочи. Данные пластинки эффективны для разделения высокомолекулярных и амфотерных соединений.
Вышеперечисленные сорбенты являются наиболее распространенными, но помимо этих существуют множество веществ, используемых как сорбенты. Это тальк, сульфат кальция, крахмал и т.д..
В то же время даже уже указанные сорбенты могут быть модифицированы для придания им новых сорбционных свойств (пропитка сорбентов реактивами, например AgNO3, создание пластин с обращенной фазой). Именно такое разнообразие возможных фаз при минимальных затратах позволяют использовать ТСХ для хроматографирования огромного числа веществ.
Растворители
В тонкослойной хроматографии, в качестве подвижной фазы используют либо чистые вещества (этилацетат, бензол и т.п.), либо смеси веществ (системы) в определенном соотношении.
Подбор подвижной фазы (системы) проводится по следующим правилам:
· Выбирают такую систему, в которой разделяемые компоненты имеют небольшую растворимость (если растворимость вещества высокая, то вещества будут перемещаться с фронтом, при низкой растворимости - оставаться на старте). При распределительной хроматографии или при использовании обращенных фаз, растворимость веществ должна быть выше в подвижной фазе, чем в неподвижной.
· Состав системы должен быть постоянным и легко воспроизводимым.
· Растворитель или компоненты системы не должны быть ядовитыми или дефицитными.
· Система должна полностью разделять вещества близкого строения, причем различия в Rf должно быть не менее 0,05.
· Система не должна вызывать химические изменения разделяемых компонентов.
· В выбранной системе анализируемые вещества должны иметь различные значения Rf и распределяться по всей длине хроматограммы. Желательно, чтобы значения Rf лежало в пределах 0,05-0,85.
· При выборе системы также необходимо учитывать природу разделяемых веществ. Так, при хроматографировании веществ, имеющих основные свойства система не должна обладать кислотными свойствами и наоборот.
Эти рекомендации дают предварительную оценку выбранной системы. Последнее слово все равно остается за экспериментом.
Ход работы
- выдержать хроматографическую пластину в склянке для хроматографии для удаления с нее примесей.
- высушить пластину
- при помощи капилляра нанести указанные преподавателем пробы и свидетели на стартовую линию пластины (для пробы в одну точку внести не менее 5-ти капилляров)
- поместить пластину с пробами в склянку и ждать, пока фронт растворителя дойдет до верхнего края пластины.
- извлечь пластину из склянки и высушить на воздухе
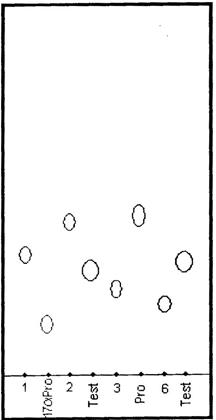
- при помощи УФ-лампы проявить хроматограмму и определить площади пятен, коэффициент разделения и коэффициент Rf.
Определяем Rf выявленных пятен по формуле:
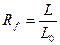 , где
, где
L – расстояние, пройденное исследуемым веществом;
L0 – расстояние, пройденное фронтом расторителя;
|