Синхронные реакции - это процессы, в которых не удается зафиксировать образование промежуточных свободных радикалов или ионов, т.е. исходная молекула сразу превращается в конечную. Поэтому подобные процессы называются еще “реакциями без механизма”. В переходных состояниях синхронных процессов присутствуют, как предполагается, частично разорванные и частично образованные связи. Существуют как мономолекулярные, так и бимолекулярные синхронные процессы.
7.1.Мономолекулярные синхронные процессы.
К ним относятся в частности, цис- и транс-изомеризация алкенов, размыкание малых циклов, распад молекул (мономолекулярное элиминирование) и сигматропные перегруппировки. Цис-транс-изомеризация алкенов возможна в результате термической или фотохимической активации:

 В переходном состоянии этой реакции, протекающей с поворотом одной половины молекулы относительно второй на 180о, происходит разрыв π-связи С=С с образованием, по-видимому, бирадикала
В переходном состоянии этой реакции, протекающей с поворотом одной половины молекулы относительно второй на 180о, происходит разрыв π-связи С=С с образованием, по-видимому, бирадикала
Энергия π-связи С=С молекулы этилена составляет около 65 ккал/моль (энергия σ-связи С-С около 83- 85 ккал/моль), поэтому термическая активация требует довольно высокой температуры (450-500о). В то же время фотохимическая активация при поглощении кванта электромагнитного излучения в ультрафиолетовой области спектра протекает достаточно легко.
 Транс-изомеры 1,2-дизамещенных этиленов более стабильны, чем цис-изомеры, ввиду стерического отталкивания заместителей в цис-изомере:
Транс-изомеры 1,2-дизамещенных этиленов более стабильны, чем цис-изомеры, ввиду стерического отталкивания заместителей в цис-изомере:
Такое отталкивание отсутствует в транс-изомере, где заместитель R расположен по разные стороны от плоскости π-связи С=С. Например, транс-бутен-2 (R=CH3) примерно на 1 ккал/моль стабильнее цис-изомера, и это различие усиливается с увеличением размера заместителя R.
Соединения с трехчленным углеродным циклом - циклопропан и его замещенные производные, хотя и обладают большой энергией углового напряжения, обусловленного тем, что углы С-С-С правильного треуго льника составляют 60о вместо нормального тетраэдрического угла sp3 -гибридизированных атомов углерода в 109.5о (энергия напряжения составляет 27.5 ккал/моль), способен к раскрытию цикла лишь при весьма высоких температурах:

В переходном состоянии этой реакции происходит, вероятно, перенос атома водорода бирадикальной частицы от среднего к крайнему атому углерода с образованием  пропена. Двойная связь С=С молекулы пропена стабилизирована эффектом гиперконъюгации с метильной группой
пропена. Двойная связь С=С молекулы пропена стабилизирована эффектом гиперконъюгации с метильной группой
и теплота образования его (-∆Hfo = 5 ккал/моль) существенно выше, чем у циклопропана.
Распад молекулы, т.е. термически инициированное мономолекулярное элиминирование в газовой фазе или в неполярных растворителях протекает, вероятно, через циклические переходные состояния:

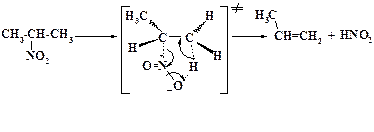
7.2 Бимолекулярные синхронные реакции носят название перициклических процессов.
Участвующие в них ненасыщенные соединения взаимодействуют с образованием циклических переходных состояний. Эти реакции подразделяются на две основные группы: циклоприсоединение – циклораспад и электроциклические реакции.
7.2.1 Циклоприсоединение – циклораспад.
Наиболее известным из процессов циклоприсоединения является реакция Дильса-Альдера, представляющая собой 1,4-присоединение алкенов к сопряженному 1,3-диену с образованием производных циклогексена:

В соответствии с принятой терминологией реакцию Дильса - Альдера относят к процессам [4+2] циклоприсоединения, т.к. в ней принимает участие 4π-электронная система диена и 2 π-электронная система алкена, который называется диенофилом. Продукт реакции Дильса-Альдера называют аддуктом. В этой реакции практически синхронно рвутся две π-связи в исходных реагентах и одновременно образуются две новые σ-связи и одна π-связь в аддукте. Поскольку σ-связи прочнее π-связей, реакции [4+2]циклоприсоединения экзотермичны, хотя многие из них обратимы. Обратный процесс, протекающий при высокой температуре, называется “ретродиеновым распадом”, он протекает через циклическое переходное состояние и также подпадает под определение перициклических реакций:
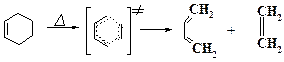
Наиболее реакционноспособными диенофилами являются алкены CH2=CH-X, у которых двойная связь сопряжена с электроноакцепторной группой X=COOR, C(=O)R, NO2, CN и т.д., а наиболее активными диенами являются диены, содержащие электронодонорные группы. При этом верхняя занятая молекулярная орбиталь донора (диена) взаимодействует с нижней вакантной МО акцептора (диенофила).
Реакция Дильса-Альдера стереоспецифична, т.е. имеет место исключительно цис-присоединение к двойной связи.
Таким образом, конфигурации диена (цис-) и диенофила (цис- или транс-) сохраняются при образовании аддукта:

 Особенно легко протекает циклоприсоединение с образованием бициклической системы (при использовании циклического диена), например,
Особенно легко протекает циклоприсоединение с образованием бициклической системы (при использовании циклического диена), например,
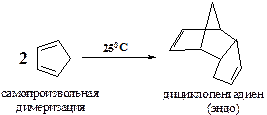
При этом преимущественно образуется изомер с транс-расположением заместителя в алкене и метиленового мостика в бициклической системе (эндо-изомер).
В качестве второго примера перициклических реакций можно привести реакцию 1,3-диполярного циклоприсоединения, или [3+2]-циклоприсоединения, где молекула, которую можно представить в виде 1,3-диполя, реагирует с этиленовым (или ацетиленовым, нитрильным и др.) фрагментом другой молекулы, называемой диполярофилом, с образованием 5-членного гетероцикла. Примеры 1,3-диполей приведены ниже:
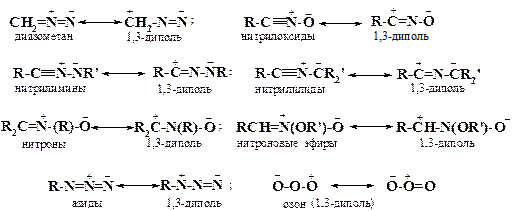
В переходном состоянии этой реакции происходит циклическая перестройка
π-электронного облака. Такое переходное состояние с круговой делокализацией электронов называется перициклом:


Простейшей реакцией циклоприсоединения является димеризация этилена с образованием циклобутана ([2+2] –циклоприсоединение). Однако, в отличие от вышеприведенных типов циклоприсоединения, она проходит только в условиях фотохимической активации при облучении ультрафиолетовым светом. Для объяснения этого факта рассмотрим характер взаимодействующих молекулярных орбиталей:

Таким образом, из двух π-связей молекул этилена образуются две σ-связи молекулы циклобутана. Эти π-орбитали называются перициклическими и играют определяющую роль в реакции. Для прохождения реакции необходимо, чтобы верхняя занятая молекулярная орбиталь (ВЗМО) донора вступала во взаимодействие (перекрывание) с нижней вакантной орбиталью (НВМО) акцептора:

Однако такое перекрывание невозможно по соображениям симметрии, т.к. перекрываться могут МО, у которых знак волновой функции одинаков в области перекрывания. Для двух молекул этилена в основном электронном состоянии это условие не соблюдается. Однако при электронном возбуждении УФ светом один электрон переходит с ВЗМО на разрыхляющую π*-орбиталь и в таком виде молекула этилена может взаимодействовать с акцептором, приводя к образованию продукта циклоприсоединения.
7.2.2 Электроциклические реакции.
Электроциклической реакцией называется циклизация путем образования σ-связи между концевыми атомами линейной сопряженной системы π-связей, а также обратный процесс разрыва этой σ-связи в циклоалкенах (циклополиенах). Как и циклоприсоединение, электроциклические реакции являются либо термически, либо фотохимиически активируемыми. Эти реакции всегда стереоспецифичны, т.е. стереоизомерные реагенты в одних и тех же условиях реакции образуют стереоизомерные продукты.
Так, цис-3,4-диметилциклобутен при нагревании до 1750С образует исключительно транс-цис(2Z,4E)-2,4-гексадиен, а транс-3,4-диметилциклобутен – транс-транс(2E,4E)-2,4-гексадиен:

При раскрытии напряженного четырехчленного цикла снимается угловое напряжение, поэтому реакция идет в сторону ациклических соединений. Однако имеются примеры и другого рода, когда более стабильным оказывается циклический продукт. Так, цис-транс-2,3-дифенил-1,4-динитробутадиен-1,3 при нагревании циклизуется до  цис-1,2-динитро-3,4-дифенилциклобутена:
цис-1,2-динитро-3,4-дифенилциклобутена:
Наблюдаемая стереоспецифичность обусловлена тем, что группы, связанные с концами сопряженной диеновой системы, поворачиваются в процессе раскрытия цикла в одном и том же направлении. Такое вращение называется конротаторным.
Обратная реакция циклизации бутадиенов в циклобутены также должна быть конротаторным процессом, но в большинстве случаев она термодинамически невыгодна.
Другим примером электроциклических реакций может служить циклизация гексатриенов в производные циклогексадиена:

В этих случаях циклизация термодинамически выгодна. Данная реакция также стереоспецифична. Так, E, Z, E- окта-2,4,6-триен циклизуется только в цис-диметилциклогексадиен, а E, Z, Z-октатриен дает исключительно транс-изомер циклогексадиена. При этом заместители на концах триеновой системы поворачиваются в процессе циклизации в противоположные стороны, т.е. дисротаторно.
Таким образом, 4π-электронные системы претерпевают конротаторное превращение, а 6 π-электронные - дисротаторное.
Электроциклические реакции подробно изучены Лауреатами Нобелевской премии 1965 г. Р.Вудвордом и Р. Хофманом. Согласно их исследованиям, стереоспецифичность электроциклических реакций объясняется тем, что они подчиняются орбитальному контролю, а именно тем, что стереохимия таких реакций определяется свойствами симметрии верхней занятой молекулярной орбитали (ВЗМО) партнера с открытой цепью. Это предположение основано на допущении, что в процессах, включающих перераспределение электронной плотности, именно электроны с высшей энергией, т.е. занимающие ВЗМО, имеют первостепенное значение. Так, для бутадиена ВЗМО имеет  следующие свойства симметрии (А):
следующие свойства симметрии (А):

Для образования связи между концами сопряженной системы положительная доля орбитали первого углеродного атома должна перекрываться с положительной долей орбитали четвертого атома (или отрицательная с отрицательной). Это перекрывание может быть достигнуто только при конротаторном вращении.
В то же время для 6π-орбитальных систем ВЗМО имеет строение (В).
В этом случае дисротаторное замыкание цикла обеспечивает перекрывание одноименных (положительных либо отрицательных) долей орбиталей С-1 и С-6. В общем случае термические (4q+2, где q= 0, 1, 2, 3….) - электронные реакции происходят дисротаторно, а термические (4r, r=1, 2, 3….) - электронные реакции протекают конротаторно Для фотохимически инициированных реакций все должно быть наоборот.
7.2.3 Сигматропные сдвиги (внутримолекулярные перегруппировки).
Перегруппировкой называется процесс перемещения мигрирующей группы (W) от одного атома молекулы к другому, которое сопровождается разрывом одной и образованием другой σ-связи:

Атом А называют начальным местом миграции, атом В – конечным местом миграции. Атомы А и В могут быть связаны друг с другом или разделены промежуточными группами, например, -CH2- или –СН=. Мигрирующая группа может быть одноатомной (например, W=H или Br) или многоатомной (Alk, Ar и т.д.)
Все перегруппировки делятся на два типа: межмолекулярные и внутримолекулярные. В первом случае группа W полностью утрачивает связь с атомом А и может присоединиться к атому В другой молекулы. Во втором случае связь W с системой А-В непрерывно сохраняется, т.е. перегруппировка протекает через циклическое переходное

состояние. Таким образом σ-связь W-B, существующая в конечном продукте, образуется синхронно с разрывом σ-связи W-A. Для этого процесса можно представить два альтернативных переходных состояния
Пунктирные линии в структурах I и II означают частично образовавшиеся и частично разорванные связи. При этом в структуре II участвующие в реакции электроны образуют циклическую систему, а в структуре I-ациклическую систему. Циклическая электронная система может быть ароматической (по Хюккелю) или антиароматической, т.е. более или менее устойчивой по сравнению с ациклической системой.
Подавляющее большинство внутримолекулярных перегруппировок протекает через ароматическое переходное состояние, например, 1,2-сдвиги в карбокатионах:

Если перегруппировка идет через ароматическое циклическое переходное состояние с полной круговой делокализацией электронов (структура II), то ее можно отнести к перициклическим реакциям. В соответствии с классификацией Вудворда- Гофмана такие процессы называют сигматропными сдвигами. В большинстве случаев сигматропные перегруппировки представляют собой миграцию σ-связи, соседней с одной или двумя сопряженными π-системами. Каждый из концов этой σ-связи в исходном состоянии обозначается цифрой 1, а конечное положение мигрируюшей σ-связи обозначается цифрами i и j. Реакция называется [i,j] –сигматропным сдвигом, если σ-связь из положения 1,1 переходит в положение [i,j]. Так, приведенная выше реакция будет называться [1,2]-сигматропным сдвигом;миграция водорода вдоль аллильной системы представляют собой [1,3]-сигматропный сдвиг:

По характеру конечного места миграции перегруппировки делятся на четыре типа:
7.2.3.1  Перегруппировки к электронодефицитному центру, которые называются нуклеофильными или анионотропными перегруппировками:
Перегруппировки к электронодефицитному центру, которые называются нуклеофильными или анионотропными перегруппировками:
Сюда относятся перегруппировки карбокатионов, карбенов и нитренов. Такие перегруппировки в электронодефицитных системах иногда называют секстетными, т.к. конечное место миграции имеет секстет электронов вместо октета. Примеры нуклеофильных перегруппировок к электронодефицитному атому азота приведены ниже.
7.2.3.2 Перегруппировка Гофмана, состоящая в реакции первичного амида с гипобромитом натрия (NaBrO) и приводящая к образованию амина, укороченного на один углеродный атом.
 Первичным продуктом перегруппировки является изоцианат, который в условиях реакции гидролизуется до спонтанно декарбоксилирующейся замещенной карбаминовой кислоты:
Первичным продуктом перегруппировки является изоцианат, который в условиях реакции гидролизуется до спонтанно декарбоксилирующейся замещенной карбаминовой кислоты:
Механизм перегруппировки Гофмана имеет вид:

Сначала амид бромируется по азоту, N-бромамид отщепляет протон под действием гидроксил-иона и претерпевает [1,2 ]-сигматропный сдвиг.
7.2.3.3 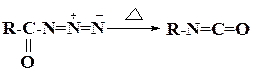 Перегруппировка Курциуса представляет собой пиролиз ацилазидов с образованием изоцианатов:
Перегруппировка Курциуса представляет собой пиролиз ацилазидов с образованием изоцианатов:
Механизм ее сходен с перегруппировкой Гофмана:

7.2.3.4  Перегруппировка Шмидта происходит при кислотно-катализируемом присоединении азотистоводородной кислоты к карбоновым кислотам, альдегидам или кетонам:
Перегруппировка Шмидта происходит при кислотно-катализируемом присоединении азотистоводородной кислоты к карбоновым кислотам, альдегидам или кетонам:
Образующийся в результате перегруппировки изоцианат, как и в реакции Гофмана, в присутствии влаги превращается в амин. Катализатором перегруппировки Шмидта чаще всего является концентрированная H2SO4, протонирующая карбоновую кислоту:

7.2.3.5 Перегруппировка Бекмана состоит в превращении оксимов в замещенные
 амиды под действием сильных кислот Бренстеда или PCl5. В большинстве случаев мигрирует
амиды под действием сильных кислот Бренстеда или PCl5. В большинстве случаев мигрирует
 группа, находящаяся в транс-положении к уходящей группе, которой при катализе кислотами Бренстеда является вода:
группа, находящаяся в транс-положении к уходящей группе, которой при катализе кислотами Бренстеда является вода:
7.2.4 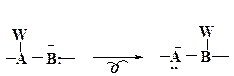 Перегруппировки к электроноизбыточному центру, или электрофильные перегруппировки:
Перегруппировки к электроноизбыточному центру, или электрофильные перегруппировки:
К ним относятся, в частности, перегруппировки Стивенса, Мейзенгеймера и Виттига.
7.2.4.1 Перегруппировка Стивенса.
При обработке сильными основаниями четвертичных солей аммония, имеющих у атома углерода, связанного с азотом, ацидифицирующую группу Z (Z=Ph, RC=O, –CN и др.), происходит перегруппировка согласно нижеприведенной схеме:
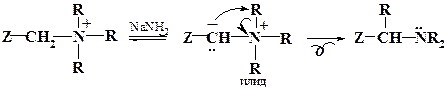
7.2.4.2 Перегруппировка Мейзенгеймера, в которой оксид третичного амина при нагревании перегруппировывается в замещенный гидроксиламин:

7.2.4.3 Перегруппировка Виттига, которую претерпевают простые эфиры при действии магнийорганических соединений, превращаясь в алкоголяты лития:

Указанные перегруппировки представляют собой 1,2-алкильные сдвиги и называются анионными 1,2-сдвигами.
 7.2.4.4 Перегруппировки свободных радикалов.
7.2.4.4 Перегруппировки свободных радикалов.
Примером 1,2-перегруппировки свободных радикалов может служить миграция атома хлора, происходящая в ходе реакции 3,3,3-трихлорпропена с Br2 в присутствии пероксидов:

7.2.4.5 Перегруппировки нейтральных π-систем.
Перегруппировки типов 7.2.3.2 - 7.2.3.5 и 7.2.4.1 - 7.2.4.4 имеют место в карбокатионах, карбанионах или свободных радикалах. Эти частицы обычно малоустойчивы и имеют короткое время жизни. Однако это не значит, что в процессе перегруппировки происходит полный разрыв связи с уходящей группой с образованием свободного катиона, аниона или радикала. В большинстве случаев мигрирующая группа начинает переходить к конечному месту миграции еще до того, как полностью сформировался ионный или радикальный центр. В связи с этим перегруппировки типов 7.2.3.2 - 7.2.3.5 и 7.2.4.1 - 7.2.4.4 называют перегруппировками в насыщенных системах.
Четвертый тип перегруппировок наблюдается в ненасыщенных π-электронных системах при термической или фотохимической активации этих весьма устойчивых в обычных условиях молекул.
Примером может служить [3,3]-сигматропная перегруппировка Коупа, в которую вступают 1,5-гексадиен и его алкильные производные при нагревании до ~300оС. Эта реакция 6-электронная и ее переходное состояние можно рассматривать как результат взаимодействия двух аллильных π-систем:

 Такое переходное состояние напоминает 6π-электронную систему бензола, но, в отличие от бензола, аллильные фрагменты смещены друг относительно друга и перекрываются не обеими, а лишь
Такое переходное состояние напоминает 6π-электронную систему бензола, но, в отличие от бензола, аллильные фрагменты смещены друг относительно друга и перекрываются не обеими, а лишь
одной долей концевых р -орбиталей:
Использованная литература:
1. В.Ф.Травень, Органическая химия, т.1,2 (учебник для Вузов). Москва, ИКЦ ”Академкнига”, 2004.
2. О.А.Реутов, А.Л.Курц. К.П.Бутин, Органическая химия, в 4-х частях, 2-ое изд., Москва, Бином. Лаборатория знаний,2005.
3. П.Сайкс, Механизмы реакций в органической химии, 4-ое изд., Москва, Химия, 1991.
ОГЛАВЛЕНИЕ
Введение…………………………………………………………………………………….3
1. Классификация механизмов реакций по характеру разрыва и
образования связи…………………………………………...............................................4
1.1.Гомолитические реакции………………………………………………………........4
1.2.Гетеролитические, или ионные реакции……………………………….………......6
1.2.1. Карбокатионы…………………………………………………………...........6
1.2.2. Катионы с зарядом на гетероатоме. ………………………………………...7
1.2.3. Карбанионы……………………………………………………………………9
1.2.4. Реакции ионов………………………………………………………………..11 1.2.5.Нуклеофильные и электрофильные реагенты и реакции…......................................14
2 Классификация гетеролитических реакций по типу превращения субстрата………...17
2.1. Реакции замещения…………………………………………………………………….17
2.2. Реакции присоединения……………………………………………………...........19
2.3. Реакции элиминирования……………………………………………….................21
2.4. Мономолекулярная фрагментация………………………………………………..24
3. Стереохимические аспекты нуклеофильного замещения……………………………..25
4. Корреляция параметров нуклеофильности…………………………………………….30
5. Особенности различных типов нуклеофильного замещения………………………...34
5.1. Анхимерное содействие…………………………………………………………34
5.2. Нуклеофильное ароматическое замещение…(SNAr)………………………….35
5.3. Анион-радикальное нуклеофильное замещение (SRN1)………………………36
5.4. Викариозное нуклеофильное замещение SNHAr)……………………………...37
5.5. Мономолекулярное нуклеофильное ароматическое замещение (SN1)………38
6. Электрофильное ароматическое замещение (SEAr)…………………………………..38
7. Синхронные (молекулярные) реакции………………………………………………...40
7.1. Мономолекулярные синхронные процессы…………………………………….40
7.2. Бимолекулярные синхронные реакции………………………………………. 42
7.2.1. Циклоприсоединение - циклораспад………………………………….42
7.2.2. Электроциклические реакции………………………………………….45
7.2.3.Сигматропные сдвиги(внутримолекулярные перегруппровки)……..47
7.2.3.1 Перегруппировки к электронодефицитному центру………………..49
7.2.4. Перегруппировки к электроноизбыточному центру………………... 51
Список литературы………………………………………………………………………. 53