КОНТРОЛЬНЫЕ ВОПРОСЫДЛЯ ПОДГОТОВКИ К ЭКЗАМЕНУ (ЗАЧЕТУ) ПО
АНАЛИТИЧЕСКОЙ ХИМИИ
1. Понятия аналитическая химия и химический анализ.
Аналитическая химия – раздел химической науки, разрабатывающей на основе фундаментальных законов химии и физики принципиальные методы и приемы качественного и количественного анализа атомного, молекулярного и фазового состава вещества
Анализ вещества (химический анализ) – получение опытным путем данных о химическом составе вещества любыми методами: физическими, химическими, физико-химическими методами.
2. Аналитические реакции. Условия выполнения реакций. Чувствительность реакций.
Реакцию, используемую для аналитических целей, называют аналитической, а вещество, ее вызывающий – реагент. Аналитический сигнал – любое свойство системы, отличающее ее от остальных.
Различают три вида реакций: реакция открытая (обнаружения), реакция идентификации (подтверждение) или проверка правильности открытия, реакция осаждения (отделения).
Аналитические реакции делят на мокрые и сухие (окрашивание пламени).
Условия выполнения реакции: среда (кислая, нейтральная, щелочная) создается прибавлением к раствору кислоту, щелочи или буферной смеси.
Температура должна быть соответствующей. Одни реакции идут при нагревании, а другие – в холоде.
Концентрация должна быть достаточной, така как осадок образуется только из перенасыщенного раствора, то есть его концентрация больше растворимости.
Чувствительность реакции характеризуется: открываемый минимум (т) – наименьшая масса вещества в мкг, которая может быть обнаружена посредством данной реакции, (т)< 50. Минимальная предельная концентрация – характеризует минимальную концентрацию Сmin, q – объем раствора в мл. Предельное разбавление – (q) – величина, обратная концентрации. Отсюда, m=Cmin * Vmin * 10^6 = 1/q * Vmin * 10^6
Чем меньше открываемый минимум и предельная концентрация и чем больше предельное разбавление, тем реакция чувствительнее.
3. Математическая обработка результатов анализа.
Получение результатов количественного химического анализа, как и любых других измерений всегда сопряжено с погрешностями. Погрешности характеру вызывающих их причин классифицируют на систематические, случайные и промахи. Плодотворность такого подхода состоит в том, что он позволяет наметить общую стратегию уменьшения погрешностей путем поэтапной борьбы с систематическими, а потом (при снижении их до уровня случайных) – со случайными. Систематические погрешности вызываются или известными причинами, или такими, которые могут быть установлены при детальном анализе процедуры измерений. Их можно выявить, устранить или учесть в окончательных расчетах. Случайные погрешности, в отличие от систематических, не имеют видимой причины. Точнее говоря, их причины столь многочисленны в своей совокупности, и каждая из причин столь незначительно влияет на результат измерений, что их индивидуальное рассмотрение не имеет смысла. Случайные погрешности можно оценить по законам математической статистики. Причем общая случайная погрешность каждого измерения не постоянна ни по значению, ни по знаку, но появление значительной случайной погрешности тем менее вероятно, чем больше её абсолютное значение. На рис. 1 показаны различные случаи измерений в зависимости от погрешностей той или иной природы.

Рис. 1. Зависимость погрешности измерений от n в экспериментальных сериях: 1 – идеальная; 2 – при отсутствии случайной и наличии постоянной систематической погрешности; 3 – в отсутствии систематической и наличие случайной погрешности; 4 –реальная.
Прямая 1 – образ идеальной серии измерений, без систематических и без случайных погрешностей (все результаты совпадают с истинным значением 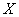 . Ломаная линия 3 – образ серии измерений, лишенных систематической погрешности (
. Ломаная линия 3 – образ серии измерений, лишенных систематической погрешности ( ), а среднее значение совпадает с прямой 1 (
), а среднее значение совпадает с прямой 1 (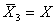 ). Прямая 2 соответствует серии измерений, не содержащих случайной, но содержащих постоянную систематическую погрешность
). Прямая 2 соответствует серии измерений, не содержащих случайной, но содержащих постоянную систематическую погрешность 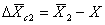 .
.
Реальной серии измерений соответствует ломаная линия 4, где мерой систематической погрешности служит разность  , а мерой случайной погрешности разность
, а мерой случайной погрешности разность 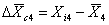 .
.
4. Растворы. Способы выражения концентрации веществ в химическом анализе: молярная, нормальная, титр раствора.

5. Гетерогенное равновесие "осадок-раствор". Молярная и массовая растворимость малорастворимых соединений.
Закон действующих масс применим как к гомогенным равновесиям, например, к равновесиям в растворах, так и к равновесиям в гетерогенных системах.
Гомогенная (однородная) система – это система, физические и химические свойства которой во всех еѐ частях одинаковы. Она состоит только из одной фазы – твердой, жидкой или газообразной. Типичным примером гомогенной системы являются растворы.
Гетерогенная (неоднородная) система – это система, состоящая из нескольких гомогенных фаз, разделенных между собой поверхностью раздела. Свойства фаз отличаются друг от друга и примерами таких гетерогенных систем могут являться две несмешивающиеся жидкости, осадок и насыщенный раствор над ним, газ и твердое вещество и др.
Равновесие, устанавливающееся в гетерогенной системе на границе раздела фаз, называется гетерогенным равновесием. Гетерогенная система «осадок – раствор» представляет собой осадок малорастворимого соединения, например, сульфата бария BaSO4, находящийся в равновесии с его насыщенным раствором: BaSO4 (т) ↔ Ba2+ + SO4 2- (р)
Напомним, что насыщенным называется раствор, содержащий максимальное количество вещества, которое может раствориться в данном объёме раствора при данной температуре и давлении. Насыщенный раствор устойчив во времени.
Пересыщенный раствор – это раствор, в котором содержится большее количество вещества, чем в насыщенном. Такой раствор неустойчив и избыток вещества выделяется из него в твердую фазу, то есть образуется осадок. Процесс протекает до образования над осадком насыщенного раствора.
Ненасыщенный раствор характеризуется меньшим количеством растворенного вещества, чем насыщенный, и в нем может быть растворено дополнительное количество вещества до получения насыщенного раствора.
Концентрация вещества в насыщенном растворе называется растворимостью. Растворимость является количественной характеристикой способности вещества растворяться. Различают молярную и массовую растворимость вещества.
Молярная растворимость вещества (S, моль/л) – это количество растворенного вещества, содержащееся в одном литре его насыщенного раствора:
S= n /V= m/ M* V
где: n – количество растворенного вещества, моль m – масса растворенного вещества, г M – молярная масса растворенного вещества, г/моль V – объѐм насыщенного раствора, л 2
Массовая растворимость вещества (Т, г/л) – это масса растворенного вещества, содержащаяся в одном литре его насыщенного раствора:
T= m/ V
где: m – масса растворенного вещества, г V – объѐм насыщенного раствора, л Молярная и массовая растворимости связаны между собой соотношением: Т = S∙M
6. Произведение растворимости.
К равновесным системам следует отнести также и систему труднорастворимый электролит – его насыщенный раствор. В этом случае мы имеем дело с динамическим гетерогенным равновесием 

|
В этом растворе концентрация ионов очень мала и вследствие этого взаимодействие их друг с другом практически отсутствует.
Константу равновесия для гетерогенной системы можно записать так:

|
концентрацию твердой фазы AgClk можно считать постоянной, тогда

|
С другой стороны,

|
Так как 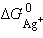 ,
, 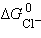 и
и  – постоянные величины, то
– постоянные величины, то
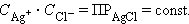
|
В общем виде для уравнения

|
|

В насыщенном растворе труднорастворимого сильного электролита произведение концентрации его ионов в степенях стехиометрических коэффициентов при данной температуре есть величина постоянная, называемая произведением растворимости (ПР).
Произведение растворимости характеризует растворимость труднорастворимого электролита при данной температуре. Из двух однотипных солей, например, CaSO4 с ПР = 2,5∙10–5 и BaSO4 с ПР = 1,1∙10–10, большей растворимостью обладает та соль, у которой ПР больше.
7. Влияние одноименного иона на растворимость малорастворимых соединений.

8. Качественный анализ. Понятие дробного и систематического качественного анализа.
Применяя специфические реакции можно обнаружить ионы так называемым дробным методом, т.е. непосредственно в отдельных порциях раздробленного раствора, не учитывая возможное присутствие других ионов, при этом не имеет значение и порядок обнаружения ионов. Если нет специфических реакций, то используют определенную последовательность реакций обнаружения отдельных ионов, которую называют систематическим анализом. Он состоит в том, что к обнаружению каждого иона приступают лишь после того, как другие ионы, мешающие его обнаружению, не будут предварительно обнаружены и удалены из раствора. В систематическом анализе ионы выделяются не поодиночке, а целыми группами. Так называемым групповым реагентом.
9. Качественный анализ катионов.
Классификация катионов (кислотно-щелочной метод)
| Хлориды, сульфаты и гид-роксиды растворимы в воде | Хлориды нераство-римы в воде и в разбав-ленных кислотах | Сульфаты нераство-римы в во-де и в кис-лотах | Гидроксиды амфотерны: растворимы в избытке щелочи | Гидроксиды нераствори-мы в избытке щелочи | Гидроксиды образуют растворимые аммины |
| I | II | III | IV | V | VI |
| K+, Na+, NH+4 | Ag+, Pb2+, Hg2+2 | Ba2+, Pb2+, Sr2+ | Al3+, Cu2+, Zn2+, Sn2+, Sn4+, As5+, As3+ | Mg2+, Mn2+, Fe2+, Fe3+, Bi3+, Sb3+, Sb5+ | Cu2+, Co2+, Ni2+, Cd2+, Hg2+ |
| Группо-вого реа-гента нет | Группо-вой реа-гент: 2 н. раствор НCl | Групповой реагент: 2 н. р-р H2SO4 | Групповой реагент: избыток 4 н. NaOH или KOH | Групповой реагент: избыток 25% - ного NH3 | Групповой реагент: избыток 25% - ного NH3 |
10. Качественный анализ анионов.
Классификация анионов
| Номер группы | Характеристика групп | Анионы, образующие группу | Групповой реагент |
| I | Соли бария мало растворимы в воде | SO42-, SO2-3, CO2-3, PO3-4, C2O2-4 и др. | BaCl2 в нейтральном или слабощелочном растворе |
| II | Соли сесребра малорастворимы в воде и разбавлен-ной HNO3 | Cl-, Br-, I-, S2-, SCN-, CN- и др. | AgNO3 в присутствии 2 н. HNO3 |
| III | Соли бария и се-ребра растворимы в воде | NO-3-, NO-2, CH3COO- и др. | Группового реагента нет |
11. Реакция обнаружения отдельных катионов. Реакции обнаружения отдельных анионов


12. Количественный химический анализ.
Задача количественного анализа — определить количество элемента или соединения а исследуемом образце. Чаще интересует не абсолютное количество, а концентрация, либо массовый процент. Следует помнить, что методы аналитической химии не рассчитаны на определение «полного точного состава». Любой метод способен определить только «концентрацию чего-то в чем-то». К примеру, метод, позволяющий определить концентрацию алюминия в воде, не подойдет для определения концентрации алюминия в краске или в пищевых продуктах. Поэтому количественный химический анализ — это заключительный этап исследования химического состава для неизвестного образца. И прежде чем провести количественный анализ, необходимо точно узнать, из каких веществ состоит образец (этап качественного анализа) и какие конкретно вещества следует определить количественно.
13. Классификация химических методов анализа.
По области применения:
а) Групповые (применяются групповые реагенты) реакции идущие с определенной группой элементов или веществ.
Применяются:
1)Для обнаружения присутствия элементов определенной аналитической группы;
2)В систематическом ходе анализа для полного отделения аналитической группы от других групп;
3)Для концентрирования групп веществ;
4)Для отделения групп веществ мешающих обнаружению искомых соединений.
б) Характерные реакции различают по селективности
1). Селективные (избирательные) – для открытия ограниченного числа ионов (веществ) (2-5) – дают с ними одинаковые или сходные аналитическое реакции;
2). Специфичные – высокоселективные – для открытия 1 компонента.
Избирательность реакций может быть повышена путем применения приемов маскирования, регулирования условий проведения (рН, температуры), выделения и разделения обнаруживаемых компонентов.
Важной характеристикой аналитических реакций, применяемых для обнаружения веществ является предел обнаружения – наименьшее содержание, которое сложно обнаружить в присутствии определяемого компонента с заданной доверительной вероятностью.
14. Гравиметрический метод анализа.
Гравиметрический анализ основан на законе сохранения массы и постоянства состава вещества.
Метод выделения.
Этот метод основан на извлечении из исследуемого вещества определяемого компонента в свободном состоянии, который затем точно взвешивают. Так, например находят массовую долю золы в твердом топливе.
Метод отгонки.
Этот метод основан на полном удалении определяемого компонента в виде летучего соединения и взвешивании остатка.
Методом отгонки определяют влажность материалов и т.д.
Метод осаждения.
Этот метод основан на количественном осаждении искомого иона в виде малорастворимого соединения определенного химического состава. Выделившийся осадок отфильтровывают, промывают, высушивают, прокаливают и точно взвешивают.
15. Требования, предъявляемые к осадкам и весовым формам в гравиметрии.
В ходе любого анализа можно выделить основные этапы:
отбор и усреднение пробы;
взятие навески;
разложение, растворение пробы;
разделение (выделение определяемого компонента и концентрирование);
количественное измерение;
запись результатов анализа и расчеты.
Осажденной формой называют соединение, в виде которого определяемый компонент осаждается из раствора. Гравиметрической (весовой) формой называют соединение, которое взвешивают.
Осажденная форма должна быть:
· достаточно малорастворимой, чтобы обеспечить практически полное выделение определяемого вещества из раствора.
· полученный осадок должен быть чистым и легко фильтрующимся (что определяет преимущества кристаллических осадков);
· осажденная форма должна легко переходить в гравиметрическую форму.
Основными требованиями к гравиметрической форме являются:
· точное соответствие ее состава определенной химической формуле;
· химическая устойчивость в достаточно широком интервале температур, отсутствие гигроскопичности;
· как можно большая молекулярная масса с наименьшим содержанием в ней определяемого компонента для уменьшения влияния погрешностей.
16. Титриметрические методы анализа, классификация методов по характеру реакции, лежащей в основе титрования.
Основан на определении объемов растворов (взаимодействующих веществ), концентрация одного из которых известна. При выполнении анализа к точно измеренному объёму анализируемого образца постепенно прибавляют непрерывно контролируемое кол-во реагента вплоть до того момента, пока кол-во молей эквивалента добавленного реагента не станет равным кол-ву молей эквивалента определяемого компонента, т.е. до точки эквивалентности.
По используемым реакциям различают:
1. Методы кислотно-основного титрования (нейтрализации)
Определяют кислоты, щелочи, гидролизующиеся соли. Используются индикаторы, изменяющие окраску в зависимости от рН среды.
2. Методы окисления-восстановления (редоксиметрия)
- перманганатометрия, рабочий раствор КМnО4 является окислителем; определяют Fe2+, NO2−, CNS−;
- йодометрия, рабочий раствор I2 – окислитель, вспомогательный раствор KI – восстановитель; определяют KMnO4, MnO2, Cl2, Na2SO3 Cu2+;
- хроматометрия, K2Cr2O7 – окислитель;
- броматометрия, KВrO3 – окислитель;
- ванадатометрия, рабочий раствор NH4VO3 – окислитель;
- цериметрия, окислитель и рабочий раствор − соединия Се (IV).
3. Методы осаждения – определяемый элемент переходит в осадок.
По применяемому рабочему раствору существует
- аргентометрия (AgNO3);
- роданометрия (NH4CNS);
- меркурометрия (Hg22+).
4. Методы комплексообразования.
Основаны на образовании комплексных ионов. Используются комплексоны (трилон Б).
17. Требования, предъявляемые к титриметрическим реакциям.
В титриметрическом анализе может быть использована не любая химическая реакция. Реакции, применяемые в титриметрии, должны удовлетворять следующим основным требованиям:
1) реакция должна протекать количественно, т.е. константа равновесия реакции должна быть достаточно велика;
2) реакция должна протекать с большой скоростью;
3) реакция не должна осложняться протеканием побочных процессов;
4) должен существовать способ определения окончания реакции.
Если реакция не удовлетворяет хотя бы одному из этих требований, она не может быть использована в титриметрическом анализе.
18. Способы титрования (прямое, обратное, заместительное)
При прямом титровании исследуемый раствор непосредственно титруют стандартным раствором.
Когда определение точки эквивалентности затруднено, пользуются методом обратного титрования: к точно отмеренному объему раствора исследуемого вещества прибавляют точно отмеренный объем стандартного раствора, взятый в избытке; избыток оттитровывают другим стандартным раствором, расчет производят по остатку.
Косвенное титрование (метод замещения) заключается в добавлении к раствору исследуемого вещества раствора другого соединения, реагирующего с ним с образованием эквивалентного количества продукта реакции, к-рый оттитровывают стандартным раствором.
19. Приготовление первичных и вторичных стандартных растворов.
Титранты (растворы с точно известной концентрацией) делятся на первичные и вторичные стандарты.
Первичный стандарт – раствор, приготовленный из стандарта (стандартного или установочного вещества). Необходимая навеска стандарта взвешивается на аналитических весах и растворяется в мерной колбе заданного объема. Первичные стандарты применяют как для обычных титриметрических определений, так и для установления точной концентрации растворов вторичных стандартов.
Вторичные стандарты (стандартизированные растворы) – растворы веществ, не являющихся стандартами. Из таких веществ готовят растворы примерно известной концентрации, а затем устанавливают их точную концентрацию (стандартизируют), оттитровывая раствором первичного стандарта.
20. Способы установления конечной точки титрования. Индикаторы. Индикаторные ошибки.
Как правило, в титриметричном анализе установить непосредственно точку эквивалентности практически невозможно. Обычно устанавливают конечную точку титрования (КТТ). Это можно сделать двумя методами: инструментальным или визуальным.
При использовании инструментальных методов КТТ устанавливается с помощью приборов, путем измерения изменения различных физических свойств титруемых растворов, например: потенциалов, оптической плотности, удельной электропроводности и т.д. При этом определяемая КТТ максимально приближена или совпадает с точкой эквивалентности.
Визуальные методы фиксирования КТТ разделяют на безиндикаторные и индикаторные.
В безиндикаторных методах КТТ определяют с появлением или исчезновением окраски титранта или раствора.
В индикаторных методам к анализируемой жидкой пробе добавляют индикатор, который придает ей определенную окраску, и дальше титруют до резкого изменения этой окраски.
| Индикатор | рН | Изменение окраски |
| Метиловый оранжевый (метилоранж) | 3,0 ÷ 4,4 | красный – желтый |
| Метиловый красный (метилрот) | 4,2 ÷ 6,2 | красный – желтый |
| Лакмус | 6,0 ÷ 8,0 | красный – синий |
| Фенолфталеин | 8,0 ÷ 10,0 | бесцветный - красный |
Индикаторные ошибки.
Водородная индикаторная погрешность может возникнуть при недотитровании сильной кислоты (в таком случае погрешность отрицательная) либо когда сильная кислота используется в качестве титранта и добавлена в избытке (положительная погрешность).
Гидроксидная погрешность может возникнуть при недотитровании сильного основания (отрицательная погрешность) либо в том случае, когда сильное основание используется в качестве титранта и добавлено в избытке (положительная погрешность).
Водородная и гидроксидная ошибки возникают в расчетах результатов титрования сильных кислот сильными основаниями (или наоборот).
Кислотная и основная ошибки возникают в расчетах результатов титрования слабых кислот или оснований.
21. Расчет массы навески исследуемой пробы и вычисление результатов титриметрических определений.
В методе отдельных навесок навеска анализируемого образца массой m*, содержащая определяемое вещество, растворяется в воде (или другом растворителе) и полученный раствор титруется раствором титранта объемом V(T) с концентрацией C(1/zT). Закон эквивалентов в этом случае имеет вид:

где m(Х) – масса определяемого вещества X в навеске.
Отсюда масса определяемого вещества X в навеске равна:
m(Х) = С(1/zТ)∙V(Т)∙М(1/zХ).
22. Расчет содержания определяемого вещества по нормальности, по титру стандартного раствора, по титру, выраженному по определяемому веществу.
v Если известны молярная концентрация эквивалента (нормальность) титранта C 2 и объем раствора V 2 в миллилитрах, израсходованный на титрование определяемого вещества, то количество вещества титранта, затраченное на реакцию, будет равно
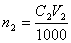 .
.
В точке эквивалентности количество вещества титранта, израсходованное на реакцию, будет точно равно количеству определяемого вещества в анализируемом растворе (n 1= n 2). Поэтому
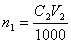 .
.
v Если известен титр T2 рабочего раствора, т.е. масса (г) растворенного вещества в 1 мл раствора, то количество вещества эквивалента титранта, вступившее в реакцию, составит
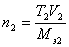 .
.
Тогда масса определяемого вещества в анализируемом растворе будет равна

 .
.
v Широко распространен в практике способ расчета результатов анализа с помощью условного титра рабочего раствора, или титра раствора по определяемому веществу T2/1. Он показывает массу определяемого вещества, которая соответствует 1 мл рабочего раствора. Поэтому масса определяемого вещества может быть рассчитана очень простым образом
m 1= T 2/1× V 2 .
Если сравнить это выражение с предыдущей формулой расчета по титру рабочего раствора, то титр по определяемому веществу можно представить в виде
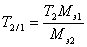 .
.
С другой стороны, из формулы расчета массы определяемого вещества через молярную концентрацию эквивалента рабочего раствора следует, что
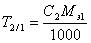
23. Расчет содержания определяемого вещества методом обратного титрования.
К определяемому веществу добавляют известный избыток титранта Т1:
X + T1 ® A + B.
Затем избыток непрореагировавшего Т1 оттитровывают титрантом Т2:
T1 + T2 ® C + D.
Закон эквивалентов для обратного титрования имеет вид:
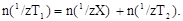
При титровании навески образца массой m(Х) закон эквивалентов принимает вид:
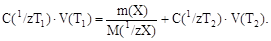
Масса определяемого вещества в навеске равна:

При титровании аликвотной доли раствора определяемого вещества V(Х) закон эквивалентов имеет вид:
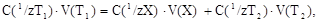
откуда рассчитывают молярную концентрацию эквивалента X:

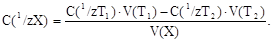
Масса определяемого вещества в объеме колбы равна:

24. Протолитическая теория кислот и оснований (теория Бренстеда-Лоури и Льюиса)
Протолитическая (протонная) теория кислот и оснований была предложена в 1923 году независимо друг от друга датским учёным Й. Брёнстедом и английским учёным Т. Лаури. В ней понятие о кислотах и основаниях было объединено в единое целое, проявляющееся в кислотно-основном взаимодействии.
Согласно этой теории кислотами являются молекулы или ионы, способные быть в данной реакции донорами протонов, а основаниями являются молекулы или ионы, присоединяющие протоны (акцепторы). Кислоты и основания получили общее название протолитов. Сущностью кислотно-основного взаимодействия является передача протона от кислоты к основанию.
25. Равновесие в водных растворах кислот и оснований.
Кислотно-основные равновесия в водных растворах. Водородный показатель рН и кислотность среды.
• Кислотно-основное (или протолитическое) равновесие – это равновесие в котором участвует протон (Н+).
• Протолитическая теория Бренстеда-Лоури:
кислоты – все частицы, способные отщеплять протон
HCN H+ + CN- HS- H+ + S 2-
основания – все частицы, способные присоединять протон
CN- + H+ HCN NН3 + H+ NH4 +
Сопряжённые кислота и основание:
HCN (к) и СN- (осн)
NН3 (осн) и NH4 + (к)
• Амфолиты (амфотерные вещества) – вещества способные как отдавать, так и принимать протоны, то есть проявлять как кислотные, так и основные свойства:
Н2О Н+ + ОН- (CN- + H2O HCN + OH-)
Н2О + Н+ Н3О+ (HCN + H2O H3O+ + CN-)
• автопротолиз воды:
Н2О + Н2О Н3О+ + ОН-
или:
Н2О Н+ + ОН-
При 250С K = 1,8·10-16, а Н2О = 55,55 моль/л, тогда:
KW = [H2O] = [H+ ][OH- ] = 10 в -14 степени
– константа автопротолиза воды или ионное произведение воды
lgKW = lg[H+ ] + lg[OH- ] = -14
-lgKW = -lg[H+ ] + -lg[OH- ] = 14
pKW = -lgKW pH = -lg[H+ ]
pOH = -lg[OH- ]
pKW = pH + pOH = 14
• Кислотность среды:
рН < 3 – раствор сильнокислый
5 < pH < 7 – раствор слабокислый
7 < pH < 9 – раствор слабощелочной
рН > 11 – раствор сильнощелочной
| Раствор | Формула для расчета рН |
| Общий случай | pН = –lg[H+] pH = 14 + lg[OH–] |
| Раствор сильной одноосновной кислоты | [H+] = С (кислоты) pН = –lg С (кислоты) |
| Раствор сильной двухосновной кислоты | [H+] = 2 С (кислоты) pН = –lg[2 С (кислоты)] |
| Раствор слабой кислоты | pН = ½р К А – ½lg С (кислоты) |
| Раствор сильного однокислотного основания | [OH–] = С (основания) pH = 14 – lg С (основания) |
| Раствор сильного двухкислотного основания | [OH–] = 2 С (основания) pH = 14 + lg[2 С (основания)] |
| Раствор слабого основания | pН = 14 – ½р К В + ½lg С (основания) |
26. Буферные растворы. Ионная сила. Буферная емкость. Расчет рН.
Буферными называют растворы, обладающие свойством достаточно стойко сохранять постоянство концентрации водородных ионов при добавле- нии к ним небольших количеств сильной кислоты или щелочи, а также при разбавлении и концентрировании. Сама способность стойко сохранять постоянное значение рН называ- ется буферным действием. Буферные системы позволяют точно регулировать концентрацию ионов водорода и гидроксид-ионов, и, следовательно, делают возможным контроль реакций, зависящих от величины рН.
Ионная сила – мера электрического взаимодействия междуу всеми ионами в растворе
Буферная емкость - это количество вещества эквивалента сильной кислоты или сильного основания, которое следует добавить к 1 л буферного раствора, чтобы изменить его рН на единицу.
| Буферный раствор | 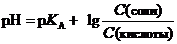

|
27. Титрование сильных и слабых одноосновных кислот и оснований.
Титрование сильной кислоты сильным основанием
Сильные кислоты и основания в воде диссоциированы полностью, поэтому концентрация ионов H+ будет равна концентрации HCl