Хромосомные болезни
Хромосомные болезни - это большая группа врожденных наследственных болезней с множественными врожденными пороками развития. В их основе лежат хромосомные или геномные мутации. Эти два разных типа мутаций для краткости объединяют термином «хромосомные аномалии».
Группы риска: может родиться ребенок с этим заболеванием, если:
1. Возраст матери старше 36 лет (здесь играют роль возрастные изменения метаболизма, что может привести к нарушению деления половых клеток во время мейоза).
2. У матери отягощенный акушерский или семейный анамнез (выкидыши, рождение детей с пороками развития и т.д.).
3. В семье уже имеются дети с хромосомными болезнями.
4. Родители контактировали с мутагенными факторами.
Хромосомные заболевания делятся на 2 группы:
1) Заболевания, связанные с аномалией аутосом:
А) числовые аномалии аутосом: синром Дауна, синдром Эдвардса, синдром Патау и т.д. (то есть ребенку через половые клетки родителей достается лишняя хромосома или нехватка хромосомы).
Б) структурные аномалии аутосом: например, синдром «крик кошки» (ребенку достается хромосома от одного из родителей с нарушением в строении).
2) Заболевания, связанные с аномалией алласом:
А) числовые аномалии: синдром Клайнфельтера, синдром Шерешевского – Тернера, синдром трипло - Х и т.д.
Б) структурные аномалии половых хромосом.
ЧИСЛОВЫЕ АНОМАЛИИ АУТОСОМ
1. Синдром Дауна – трисомия по 21 паре хромосом (47, ХХ (ХY) 21+)
Впервые синдром Дауна описал английский педиатр Л. Даун в 1866 г. Клиническая симптоматика синдрома Дауна разнообразна: от врожденных пороков развития и отклонений в умственном развитии до вторичного иммунодефицита. Детям с синдромом Дауна требуется дополнительная медицинская помощь со стороны различных специалистов, в связи с чем они составляют особую категорию в педиатрии.
Клиническая симптоматика синдрома Дауна разнообразна: это и врожденные пороки развития, и нарушения постнатального развития нервной системы, и вторичный иммунодефицит и т.п. Дети с синдромом Дауна рождаются в срок, но с умеренно выраженной пренатальной гипоплазией (на 8-10% ниже средних величин). Многие симптомы синдрома Дауна заметны уже при рождении и в последующем проявляются более четко. Квалифицированный педиатр устанавливает правильный диагноз синдрома Дауна в родильном доме не менее чем в 90% случаев. Из черепно-лицевых дизморфий отмечаются монголоидный разрез глаз (по этой причине синдром Дауна долго называли монголоидизмом), брахицефалия, круглое уплощенное лицо, плоская спинка носа, эпикант (вертикальная складка кожи, прикрывающей внутренний угол глаза), крупный (обычно высунутый) язык, деформированные ушные раковины. Мышечная гипотония сочетается с разболтанностью суставов. Часто встречаются врожденный порок сердца, клинодактилия, типичные изменения дерматоглифики (четырехпальцевая, или «обезьянья», складка на ладони), две кожные складки вместо трех на мизинце. Пороки ЖКТ наблюдаются редко.

Дети разного возраста с характерными чертами синдрома Дауна (брахицефалия, круглое лицо, макроглоссия (большой язык) и открытый рот, эпикант, гипертелоризм, широкая переносица, «карпий рот», косоглазие).

Резкая гипотония у пациента с синдромом Дауна.
Дети с синдромом Дауна относятся к часто болеющим; они тяжелее переносят детские
инфекции, чаще страдают пневмониями, средними отитами, ОРВИ, аденоидами, тонзиллитом. Слабый иммунитет и врожденные пороки являются наиболее вероятной причиной гибели детей в первые 5 лет жизни.
У большинства больных с синдромом Дауна имеются нарушения интеллектуального развития - как правило, умственная отсталость легкой или средней степени.
Моторное развитие детей с синдромом Дауна отстает от сверстников; имеет место системное недоразвитие речи. Пациенты с синдромом Дауна склонны к развитию ожирения, запоров, гипотиреоза, гнездной алопеции (облысение), рака яичка, раннему началу болезни Альцгеймера и др. Мужчины с синдромом Дауна, как правило, бесплодны; фертильность женщин заметно снижена ввиду ановуляторных циклов. Рост взрослых больных обычно на 20 см ниже среднего. Продолжительность жизни составляет около 50-60 лет.
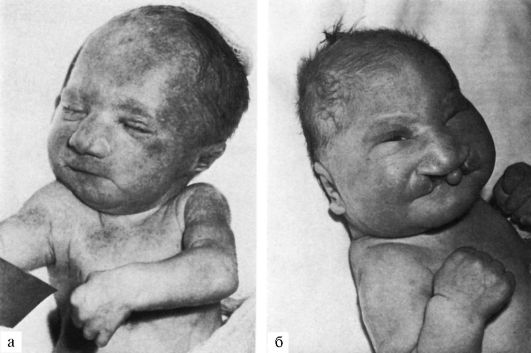
2. Синдром Патау – трисомия по 13 паре хромосом (47, ХХ (ХY) 13+)
Синдром Патау сопровождается множественными врожденными пороками развития головного мозга и лица. Это патогенетически единая группа ранних (и, следовательно, тяжелых) нарушений формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Типичный признак синдрома Патау - расщелины верхней губы и нёба (обычно двусторонние). Всегда обнаруживаются пороки нескольких внутренних органов в разных комбинациях: дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдаются полидактилия (чаще двусторонняя и на руках) и флексорное положение кистей.
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау умирают в первые недели или месяцы жизни (95% умирают до 1 года).
Лечебная помощь детям с синдромом Патау неспецифическая: операции по поводу врожденных пороков развития (по жизненным показаниям), общеукрепляющее лечение, тщательный уход, профилактика простудных и инфекционных болезней. Дети с синдромом Патау практически всегда глубокие идиоты.
3. Синдром Эдвардса – трисомия по 18 паре хромосом (47, ХХ (Х Y) 18+)
При синдроме Эдвардса отмечается выраженная задержка пренатального развития при нормальной продолжительности беременности (роды в срок). Множественные врожденные пороки развития лицевой части черепа, сердца, костной системы, половых органов. Череп долихоцефалической формы; нижняя челюсть и отверстие рта маленькие (затруднен прием пищи); глазные щели узкие и короткие; ушные раковины деформированные и низко расположенные. Из других внешних признаков отмечаются флексорное положение кистей, аномальная стопа (пятка выступает, свод провисает), I палец стоп короче II пальца. Спинномозговая грыжа и расщелина губы встречаются редко (5% случаев синдрома Эдвардса).

Новорожденный с синдромом Эдвардса (выступающий затылок, флексорное положение кисти)

Характерное для синдрома Эдвардса положение пальцев!!!

Стопа-качалка (пятка выступает, свод провисает).
Дети с синдромом Эдвардса умирают в раннем возрасте (90% до 1 года) от осложнений, обусловленных врожденными пороками развития (асфиксия, пневмония, кишечная непроходимость, сердечно-сосудистая недостаточность).