Электрохимические методы анализа, их теоретические основы и классификация
Электpохимические методы анализа (ЭМА) основаны на процессах, пpотекающих на электpодах или межэлектpодном пpостpанстве. ЭМА являются одними из стаpейших ФХМА (некотоpые описаны в конце 19 века). Их достоинством является высокая точность и сpавнительная пpостота как обоpудования, так и методик анализа. Высокая точность опpеделяется весьма точными закономеpностями используемыми в ЭМА, напpимеp, закон Фаpадея. Большим удобством является то, что в ЭМА используют электpические воздействия, и то, что pезультат этого воздействия (отклик) тоже получается в виде электрического сигнала. Это обеспечивает высокую скоpость и точность отсчета, откpывает шиpокие возможности для автоматизации. ЭМА отличаются хорошей чувствительностью и селективностью, в pяде случаев их можно отнести к микpоанализу, так как для анализа иногда достаточно менее 1 мл pаствоpа.
Инстpументом для ЭМА служит электpохимическая ячейка, пpедставляющая собой сосуд с pаствоpом электpолита, в котоpый погpужены как минимум два электpода. В зависимости от решаемой задачи pазличными могут быть фоpма и матеpиал сосуда, число и пpиpода электpодов, pаствоpа, условия анализа (пpилагаемое напpяжение (ток) и регистрируемый аналитический сигнал, температура, перемешивание, продувка инертным газом и т.п.). Опpеделяемое вещество может входить как в состав электpолита, заполняющего ячейку, так и в состав одного из электpодов. Если аналитическая окислительно-восстановительная реакция протекает на электродах ячейки самопроизвольно, то есть без приложения напряжения от внешнего источника, а только за счет разности потенциалов (ЭДС) ее электродов, то такую ячейку называют гальваническим элементом. При необходимости ячейку можно подсоединить к внешнему источнику напряжения. В этом случае, приложив достаточное напряжение, можно изменить направление окислительно-восстановительной реакции и тока на противоположное тому, что имеет место в гальваническом элементе. Окислительно-восстановительную реакцию, протекающую на электродах под действием внешнего источника напряжения, называют электролизом, а электрохимическую ячейку, являющуюся потребителем энергии, необходимой для протекания в ней химической реакции, называют электролитической ячейкой.
Полная электрическая цепь прибора для ЭМА состоит из внутренней цепи (электрохимической ячейки) и внешней цепи, включающей проводники, регуляторы тока (напряжения) и измерительные приборы.
По разновидностям аналитического сигнала ЭМА подразделяют на:
1) кондуктометрию - измерение электропроводности исследуемого раствора;
2) потенциометрию - измерение бестокового равновесного потенциала индикаторного электрода, для которого исследуемое вещество является потенциоопределяющим;
3) кулонометрию - измерение количества электричества, необходимого для полного превращения (окисления или восстановления) исследуемого вещества;
3) вольтамперометрию - измерение стационарных или нестационарных поляризационных характеристик электродов в реакциях с участием исследуемого вещества;
5) электрогравиметрию - измерение массы вещества, выделенного из раствора при электролизе.
ЭМА можно подразделить по признаку применения электролиза. На принципах электролиза базируются кулонометрия, вольтамперометрия и электрогравиметрия; электролиз не используют в кондуктометрии и потенциометрии.
ЭМА имеют самостоятельное значение для прямого проведения химического анализа, но могут применяться как вспомогательные в других методах анализа. Например, использоваться в титриметрии для регистрации конца титрования не с помощью химического цветопеременного индикатора, а по изменению потенциала, электрической проводимости тока и т.д.
Теоретические основы ЭМА
Электрод представляет собой систему, в простейшем случае состоящую из двух фаз, из которых твердая обладает электронной, а другая - жидкая - ионной проводимостью. Твердая фаза с электронной проводимостью считается проводником I рода, а жидкая фаза с ионной проводимостью - II рода. При соприкосновении этих двух проводников происходит образование двойного электрического слоя (ДЭС). Он может быть результатом обмена ионами между твердой и жидкой фазами, или результатом специфической адсорбции катионов или анионов на поверхности твердой фазы при погружении ее в воду или раствор.
При ионном механизме образования ДЭС, например в случае когда химический потенциал атомов на поверхности металла (твердой фазы) больше химического потенциала ионов в растворе, то атомы с поверхности металла будут переходить в раствор в виде катионов: Me Mez+ + ze-. Освободившиеся электроны при этом заряжают поверхность твердой фазы отрицательно и за счет этого притягивают к поверхности положительно заряженные ионы раствора. В результате на границе раздела фаз образуются два противоположно заряженных слоя, являющихся как бы обкладками своеобразного конденсатора. Для дальнейшего перехода заряженных частиц из одной фазы в другую им необходимо совершить работу, равную разности потенциалов обкладок этого конденсатора. В случае, если химический потенциал атомов на поверхности твердой фазы меньше химического потенциала ионов в растворе, то катионы из раствора переходят на поверхность твердой фазы, заряжая ее положительно: Mez++ze-Me. Как в первом, так и во втором случае указанные процессы протекают не бесконечно, а до установления динамического равновесия, которое можно изобразить обратимым редоксипереходом типа Мe -  Мez+ или в общем случае Ох +
Мez+ или в общем случае Ох +  Redz+.
Redz+.
Процессы, при которых отдача или присоединение электронов происходит на электродах, называются электродными.
Нернстом была получена формула, связывающая разность внутренних потенциалов ДЭС с активностями (концентрациями) частиц, участвующих в обратимом редоксипереходе:
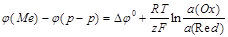 ,
,
где (Me) - потенциал заряженного слоя твердой фазы;
(раствор) - потенциал прилегающего к твердой фазе слоя раствора;
0 - константа, равная разности (Me) - (р-р), при  (Ох) = (Red) = 1 моль/л;
(Ох) = (Red) = 1 моль/л;
R - универсальная газовая постоянная (8,31 Дж/К моль);
T - температура, К;
F - число Фарадея (96 488 Кл/моль);
Z - число электронов, участвующих в редоксипереходе; 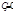 (Ох) и
(Ох) и  (Red) - активности окисленной (Ох) и восстановленной (Red) форм вещества в редоксипереходе, моль/л.
(Red) - активности окисленной (Ох) и восстановленной (Red) форм вещества в редоксипереходе, моль/л.
Установить внутренние потенциалы отдельных фаз (Me) и (р - р), к сожалению, экспериментально нельзя. Любая попытка подключить раствор с помощью провода к измерительному устройству, вызывает появление новой поверхности соприкосновения фаз металл-раствор, то есть возникновение нового электрода со своей разностью потенциалов, влияющей на измеряемую.
Однако можно измерить разность (Me) - (р - р) с помощью гальванического элемента. Гальваническим элементом называется система, составленная из двух разных электродов, обладающая способностью самопроизвольно преобразовывать химическую энергию протекающей в нем окислительно-восстановительной реакции в электрическую энергию. Электроды, из которых составлен гальванический элемент, называются полуэлементами. Протекающая в гальваническом элементе окислительно-восстановительная реакция пространственно разделена. Полуреакция окисления протекает на полуэлементе, называемом анодом (отрицательно заряженном электроде), а полуреакция восстановления - на катоде.
Электродвижущая сила (ЭДС) гальванического элемента алгебраически складывается из разностей внутренних потенциалов составляющих его электродов. Поэтому, если в качестве одного полуэлемента взять электрод с известной величиной разности внутренних потенциалов (Me) - (раствор), то по измеренной величине ЭДС можно вычислить искомую разность потенциалов исследуемого электрода.
Для этой цели принято использовать стандартный (нормальный) водородный электрод (см. рис. 1). Он состоит из платиновой пластинки или проволоки, покрытой платиновой чернью (мелкодисперсной платиной), погруженной в раствор кислоты с 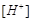 =1моль/л, давление водорода над которым 0,1 МПа (1 атм). Под каталитическим влиянием платиновой черни в электроде осуществляется обратимый редоксипереход
=1моль/л, давление водорода над которым 0,1 МПа (1 атм). Под каталитическим влиянием платиновой черни в электроде осуществляется обратимый редоксипереход  . Разность внутренних потенциалов для водородного электрода в соответствии с формулой Нернста равна:
. Разность внутренних потенциалов для водородного электрода в соответствии с формулой Нернста равна:

Рис. 1. Схема стандартного водородного электрода
 ;
;
так как [H+] = 1моль/л, а р(H2) = 1атм, то
(Me) - (р - р) = 
Решением ИЮПАК условно принято считать величину  = 0,00 В. Очевидно, что в этом случае измеренная величина ЭДС гальванического элемента, в состав которого входит водородный электрод, равна разности внутренних потенциалов второго электрода. Эту ЭДС принято называть электродным потенциалом или редоксипотенциалом и обозначать буквой Е. Переход от внутренних потенциалов к редоксипотенциалам не меняет характера формулы Нернста:
= 0,00 В. Очевидно, что в этом случае измеренная величина ЭДС гальванического элемента, в состав которого входит водородный электрод, равна разности внутренних потенциалов второго электрода. Эту ЭДС принято называть электродным потенциалом или редоксипотенциалом и обозначать буквой Е. Переход от внутренних потенциалов к редоксипотенциалам не меняет характера формулы Нернста:
 .
.
Для большинства электродов величина электродного потенциала при единичных активностях окисленной и восстановленной форм (Е0) измерена и приведена в справочниках.
При нормальных условиях и переходе от натуральных к десятичным логарифмам предлогарифмический множитель становится равным 0,0591, и формула приобретает вид
 .
.
Следует помнить, что формула Нернста связывает равновесный потенциал с активностями (концентрациями) редоксипары, т.е. потенциал, который приобретает изолированный электрод. Поэтому для аналитических цепей измерение потенциала электрода должно проводиться в условиях, максимально приближенных к равновесным: при отсутствии тока во внешней цепи гальванического элемента и через время, достаточное для достижения равновесия. Однако в реальных условиях ток может протекать через электроды. Например, ток протекает через электроды в гальваническом элементе, работа которого связана с переходом заряженных частиц через границу раздела "раствор-твердая фаза", а это направленное движение частиц есть ток. Ток протекает через электроды при электролизе, под которым подразумевают совокупность окислительно-восстановительных процессов, протекающих на электродах в растворах и расплавах электродах электролитов под действием внешнего электрического тока. При электролизе можно осуществить процессы, противоположные протекающим в гальваническом элементе.
При протекании тока (i) через электрод потенциал его изменяется и приобретает некую величину Еi, отличную от потенциала электрода в равновесных (изолированных) условиях Ер. Процесс смещения потенциала от Ер до Еi и разность Еi-Ep называют поляризацией =Ei-Ep.
Процессам поляризации подвержены не все электроды. Электроды, потенциал которых не изменяется при протекании через них тока, называют не поляризуемыми, а электроды, для которых свойственна поляризация, называют поляризуемыми.
К не поляризуемым относятся, например, электроды II рода, к поляризуемым - все металлические и амальгамные.
В качестве ионитов используют природные или синтетические, твердые, нерастворимые в воде неорганические и органические высокомолекулярные кислоты, основания и их соли, содержащие в своем составе активные (ионогенные) группы. Иониты делятся на катиониты и аниониты.
Катиониты - сорбенты, способные к обмену катионами. катиониты содержат в своем составе ионогенные группы различной степени кислотности, например сульфогруппу - SO3H, карбоксильную группу - COOH, ион водорода которых способен к катионному обмену.
Химическую формулу катионитов схематично изображают RSO3-H+, RSO3-Na+ или просто [R] H, [R] Na, где R - сложный органический радикал. Наиболее часто применяются сильнокислотные катиониты марок КУ-1, КУ-2, СДВ-2 и др.
Схема катионного обмена:
[R] H + Ме+ [R] Ме + H+
Аниониты - сорбенты, способные к обмену анионами.
Аниониты содержат в своем составе основные ионогенные группы, например, аминогруппы различной степени замещения: - NH2, =NH, N, = NH2OH, NH OH, способные к обмену гидроксид-ионов на различные анионы. Формулы анионитов схематично изображают: RNH3+OH -, RNH3+Cl - или просто [R] OH, [R] Cl. Cхема анионного обмена:
[R] OH+A - [R] A+ OH -
Применяют аниониты марок АВ-17, АН-1, ЭДЭ-10 и др.
Существуют также амфотерные иониты - сорбенты, способные как к катионному, так и к анионному обмену.
Поглощение ионов зависит от природы и структуры ионита, природы анализируемых веществ, условий проведения эксперимента (температуры, pH и др.). Каждый ионит способен поглощать определенное количество ионов, т.е. обладает определенной емкостью. Различают статическую обменную емкость (СОЕ) - количество ммоль эквивалентов иона, поглощенного за определенное время 1 г сухого ионита, и динамическую обменную емкость (ДОЕ) - количество эквивалентов ионов, поглощенных слоем ионита высотой 20 см и поперечным сечением 1 см2 при скорости пропускания 0,5 дм3/ч.
Эффект поглощения данного иона характеризуется коэффициентом распределения
Красп =  ,
,
где Сионит и Ср-р - равновесные концентрации ионов в соответствующих фазах; m - масса ионита; г; V - объем водной фазы, см3.
Ионный обмен является физико-химическим процессом, поэтому на коэффициент разделения влияют как химические, так и чисто физические факторы.
К химическим относятся следующие факторы: рН раствора, природа разделяемых ионов, их концентрация в растворе, склонность к гидратации, химический состав ионита и т.д. Например, с увеличением рН катионит увеличивает обменную емкость, а анионит - уменьшает.
К физическим факторам относятся: скорость протекания раствора через колонку, размер зерен ионита, высота колонки, температура раствора и т.д.
Для достижения оптимального разделения существенно подобрать необходимое количество ионита. Если известна константа распределения Красп и емкость данного ионита Q, то величина отношения массы ионита (m, г) к объему анализируемого раствора (V, см3), которая обеспечит уменьшение концентрации иона Меn+ в растворе от начальной величины Сн до требуемого значения Ск,
 .
.
Перед анализом ионообменную колонку регенерируют, т.е. переводят заполняющий ее ионит в определенную ионообменную форму. Зарядка катионита Н+ ионами, а анионита ОН ионами проводится путем пропускания через колонку определенного количества кислоты или основания. Затем ионит отмывают водой от избытка кислоты или основания и пропускают через него с определенной скоростью анализируемый раствор. Колонку промывают водой или другим элюентом, собирая элюат целиком или по фракциям. Ионы, поглощенные ионитом, могут быть элюированы соответствующим растворителем. Катионы, как правило, элюируют кислотой:
[R] Me + H+ [R] H + Me+;
а анионы - щелочью:
[R] A + OH [R] OH +A.
Ионообменную хроматографию применяют в следующих случаях:
· для разделения компонентов анализируемой смеси, отделения катионов и анионов, разделения катионов, разделения анионов и т.д. Например, при добавлении к смеси ионов Cu2+, Zn2+, Cd2+, Pb2+, Bi3+ соляной кислоты образуются хлоридные комплексы [CuCl4] 2-, [ZnCl4] 2-, [CdCl4] 2-, [PbCl3] - [BiCl4] -, стойкость которых растет от Cu к Bi. При пропускании через анионитную колонку комплексы поглощаются. Далее последовательно вымывают металлы разбавленной HCl, H2O и HNO3: 2-молярным раствором HCl вымывают Cu, 0.6 М HCl - Zn, 0.3М HCl - Cd, H2O - Pb, HNO3 - Bi;
· для получения аналитических концентратов. при пропускании больших объемов разбавленных растворов через слой ионита и последующем извлечении поглощенного вещества малым объемом растворителя возможно повышение концентрации вещества в 200-500 раз;
· для обнаружения ионов. Разработаны методы выделения и обнаружения всех наиболее важных ионов.
Гельхроматография - это совершенно своеобразный вид хроматографии, основанный на использовании различия в размерах молекул разделяемых веществ. Метод называют также гельфильтрационным или ситовым. НФ является растворитель, находящийся в порах геля. Гелем называют студнеобразные коллоидные растворы, в которых разбухшие частицы твердой фазы равномерно распределены в жидкой фазе.
Гель готовят на основе природных (крахмал, агар-агар) или синтетических (декстран, полиакриламид и др.) соединений.
В процессе гельхроматографирования могут быть отделены мелкие частицы, способные проникать в поры геля, от крупных. Меняя состав растворителя, можно менять степень набухания твердой фазы и, следовательно, размеры пор геля, что позволяет проводить тонкие разделения смесей.