17.1 Классификация методов анализа
Задачи идентификации (обнаружения), а также количественной оценки содержания того или иного вещества в образце решаются в рамках аналитической химии. В зависимости от цели анализа различают качественный и количественный анализ.
Целью качественного анализа является химическая идентификация, под которой понимается установление вида и состояния фаз, вида молекул, атомов, ионов и других составных частей вещества на основе сопоставления экспериментальных и соответствующих справочных данных для известных веществ.
При идентификации определяется одно, несколько, либо целый комплекс свойств веществ, например, цвет, фазовое состояние, плотность, вязкость, температуры кипения, плавления, фазового перехода, растворимость, электродный потенциал, энергия ионизации и другие свойства. Для облегчения идентификации созданы банки химических и физико-химических данных. С целью экономии времени специалистов, и упрощения доступа к обширной справочной химико-аналитической информации в настоящее время широко используются компьютеры, которыми часто снабжаются современные модели универсальных аналитических приборов, таких как спектрометры и спектрофотометры, хроматографы, полярографы и другие. Это позволяет автоматизировать как сам анализ, так и обработку его результатов.
Количественной характеристикой методик качественного анализа является предел обнаружения(открываемый минимум). Это минимальное массовое количество надежно идентифицируемого вещества, чаще всего выражаемое в мкг.
Для растворов используют величину предельной концентрации Сx,min. Если предельная концентрация выражается в г/мл, то она связана с пределом обнаружения соотношением
|
|
 . (17.1)
. (17.1)
Возможно также использование такой характеристики как предельное разбавление Dx. Предельное разбавление обратно предельной концентрации раствора и равно предельному объему раствора, который приходится на 1 мкг определяемого компонента.
Если качественный анализ проводится химическим методом, то протекающие при этом химические реакции должны быть достаточно чувствительны и специфичны. Реакция тем чувствительнее, чем меньшее количество анализируемого вещества может быть открыто в данных условиях, то есть чем меньше предел обнаружения вещества. Специфичность реакции тем выше, чем меньшее число веществ или ионов дают эту реакцию. Специфические реакции позволяют обнаружить то или иное вещество или ион в присутствии других веществ или ионов.
Примером специфических реакций может служить обнаружение ионов NH4+ в результате взаимодействия NH4++NaOH=NH3+H2O+Na+(при нагревании) по характерному запаху аммиака или посинению влажной красной лакмусовой бумажки, поднесенной к отверстию пробирки. Эти признаки являются аналитическими сигналами.
Специфична также реакция иода с крахмалом, в результате которой образуется темно-синее окрашивание.
При помощи специфических реакций можно открывать соответствующие вещества или ионы непосредственно в отдельных пробах исследуемого образца. Такая процедура носит название дробного хода анализа.
Если реакции обнаружения вещества (иона) не являются специфическими, то мешающие идентификации компоненты пробы предварительно удаляют. Например, мешающие ионы связывают в удаляемый осадок, либо в малодиссоциирующее вещество или комплексное соединение.
|
|
Анализ неизвестного вещества (иона), проводимый в определенной последовательности, при которой оно (он) идентифицируется после обнаружения и удаления мешающих анализу веществ (ионов), называется систематическим ходом анализа.
Определение содержания (концентрации, массы и т. д.) компонентов в анализируемом образце является целью количественного анализа. При количественном анализе измеряют те или иные химические, физико-химические и физические параметры анализируемого образца, которые зависят от его состава и содержания того или иного компонента. Количественный анализ поводят в определенной последовательности, в которую входят: отбор и подготовка проб, проведение анализа, обработка и расчет результатов анализа.
Продолжим рассмотрение классификации методов анализа.
В зависимости от природы анализируемого объекта различают анализ органических веществ и анализ неорганических веществ.
В зависимости от типа обнаруживаемого компонента выделяют элементный, молекулярный, изотопный, фазовый, структурно-групповой (в том числе функциональный) анализ.
По количеству взятой для анализа пробы различают макрометод анализа (берут 0,5 – 10 г сухого вещества или 10 – 100 мл раствора); полумикрометод (берут 10 – 50 мг сухого вещества или 1 – 5 мл раствора); микрометод (берут 1 – 5 мг сухого вещества или 0,1 – 0,5 мл раствора); ультрамикрометод (берут менее 1 мг сухого вещества или 0,1 мл раствора).
|
|
Методы анализа в зависимости от характера экспериментальной техники подразделяются на химические, физико-химические, физические и биологические. Биологические методы анализа используют отклик живых организмов на изменения в окружающей среде. Далее будет дан краткий и не претендующий на полноту обзор химических, физических и физико-химических методов анализа, позволяющий, тем не менее, получить общее представление об их сути.
17.2 Химические методы анализа
Эти методы основаны на протекании химических реакций. К количественным методам химического анализа относятся: гравиметрический (весовой) анализ, в котором о количестве определяемого компонента судят по массе продукта реакции, и объемный анализ. В объемном анализе о количестве определяемого компонента судят по объему жидких, твердых и газообразных продуктов реакции или реагентов, вступающих в реакцию. Познакомимся кратко с сущностью некоторых методов объемного анализа.
Объемный анализ подразделяется на газовый объемный анализ (количество анализируемого компонента определяют по объему газа, образующегося или расходующегося в результате реакции); седиментационный объемный анализ (основан на расслоении дисперсных систем под действием силы тяжести с последующим измерением объема осадка) и объемный титриметрический анализ.
В объемном титриметрическом методе анализа количество определяемого компонента устанавливается по точно измеренному объему раствора реагента, израсходованного на реакцию с анализируемым компонентом, причем концентрация раствора реагента известна с большой точностью.
Определение проводят способом титрования – это постепенное прибавление к анализируемому раствору раствора реагента точно известной концентрации (титрованный или стандартный раствор, называемый также рабочим раствором или титрантом) в количестве, эквивалентном содержанию определяемого вещества в анализируемом растворе.
Момент титрования, когда количество добавленного титранта химически эквивалентно количеству анализируемого (титруемого) вещества, называется точкой эквивалентности. Точка эквивалентности может быть установлена визуально по изменению окраски индикатора, прибавляемого в титруемый раствор.
В зависимости от характера протекающих при титровании реакций существует ряд методов титриметрического анализа. Мы остановимся на кратком рассмотрении метода кислотно-основного титрования. В основе его лежит реакция нейтрализации
Н+ + ОН – = Н2О.
В простейшем случае, при титровании сильной кислоты сильным основанием или наоборот в точке эквивалентности раствор нейтрален (рН =7).
Зависимость рН титруемого раствора от объема прилитого титранта можно выразить при помощи интегральных кривых титрования. Они могут быть получены либо в результате теоретических расчетов, либо в результате экспериментальных измерений рН при помощи прибора рН -метра.
Такая кривая для случая титрования сильной кислоты сильным основанием представлена на рисунке 17.1. Если титруется 10 мл раствора, то 1% избытка кислоты или щелочи соответствует 0,1 мл. Принимая это во внимание, из рисунка 17.1 следует, что вблизи точки эквивалентности последняя капля титранта вызывает резкое изменение рН. Этот момент в титровании получил название скачка титрования, ему соответствует вертикальный участок кривой титрования.
За ходом титрования удобно наблюдать визуально по изменению окраски кислотно-основных индикаторов. Это вещества (чаще всего слабые органические кислоты или основания), у которых при изменении рН среды изменяется окраска.
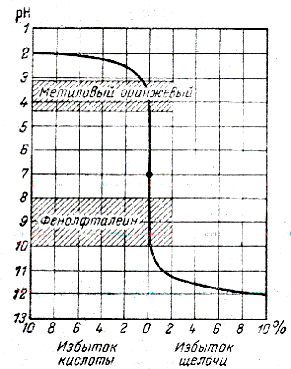
Рисунок 17.1 – Кривая титрования 0,1 н. раствора HCl 0,1 н. раствором NaOH
Интервал значений рН, в котором индикатор изменяет свою окраску, называется интервалом перехода индикатора. Для различных индикаторов он различен, так как зависит от константы диссоциации индикатора, а она определяется природой вещества и для различных индикаторов имеет различное значение. Например, метиловый оранжевый изменяет окраску от красной до желтой в интервале рН от 3,1 до 4,4; для лакмуса интервал перехода окраски от красной до синей лежит в области значений рН от 6,0 до 8,0; фенолфталеин изменяет окраску от бесцветной до малиновой в интервале рН от 8,2 до 10,0.
Для визуального фиксирования точки эквивалентности при титровании сильной кислоты сильным основанием (или наоборот) можно использовать только те кислотно-основные индикаторы, интервалы перехода которых лежат в области скачка титрования. В данном случае подойдут и фенолфталеин, и метиловый оранжевый, и лакмус.
Расчеты в методе кислотно-основного титрования основаны на законе эквивалентов. В точке эквивалентности количества вещества эквивалентов титруемого компонента и титранта равны:
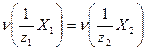 . (17.2)
. (17.2)
Если учесть, что
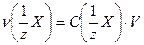 , (17.3)
, (17.3)
где 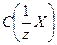 – молярная концентрация эквивалента или нормальность раствора;
– молярная концентрация эквивалента или нормальность раствора;
V – объем раствора,
то в точке эквивалентности справедливо выражение
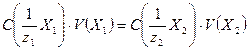 . (17.4)
. (17.4)
Иными словами, в точке эквивалентности произведения нормальностей реагирующих растворов на их объемы равны между собой. Зная объем анализируемого раствора и объем титранта точно известной нормальности, пошедший на титрование, можно, воспользовавшись формулой (17.4), легко вычислить нормальную концентрацию анализируемого раствора, то есть определить количественное содержание вещества.
17.3 Физические и физико-химические методы анализа
Физические методы анализа основываются на определении состава и содержания вещества путем измерения физических свойств, не прибегая к химическим реакциям.
Физико-химические методы базируются на использовании химических реакций, протекание которых сопровождается изменением физических свойств системы, например, изменением цвета и интенсивности окраски, величины электропроводности, теплопроводности и других свойств, зависящих от содержания вещества. Измеряя эти свойства системы, можно определить содержание анализируемого компонента.
Часто физические и физико-химические методы анализа причисляют к инструментальным методам. По сравнению с химическими методами они обладают многими достоинствами: высокой чувствительностью, возможностью одновременного определения нескольких компонентов, возможностью автоматизации и использования компьютеров, как правило, требуют меньших затрат времени.
Инструментальные методы анализа подразделяются на электрохимические, спектральные, хроматографические, радиометрические, масс-спектрометрические и другие методы.
К наиболее широко применяемым электрохимическим методам относятся потенциометрия, полярография, кондуктометрия.
Потенциометрия основана на измерении ЭДС гальванического элемента, составленного из электрода, потенциал которого при других постоянных условиях измерения зависит от концентрации соответствующих ионов в растворе, и электрода сравнения, потенциал которого не зависит от концентрации определяемого иона, погруженных в анализируемый раствор. Приборы, применяемые для подобных измерений, называются потенциометрами.
Частным случаем потенциометрии является определение рН раствора при помощи прибора рН -метра. Определение рН раствора основано на измерении ЭДС гальванического элемента, составленного из стеклянного электрода (его потенциал зависит от рН среды) и чаще всего хлорсеребряного электрода, помещенных в исследуемый раствор. ЭДС системы регистрируется милливольтметром. Так как ЭДС зависит от рН раствора, то шкалу прибора градуируют непосредственно в единицах рН.
Потенциометрию применяют в титриметрических определениях, измеряя изменяющуюся в ходе реакции ЭДС. Точку эквивалентности определяют по резкому изменению ЭДС системы.
В полярографическом анализе о количестве определяемого иона в растворе судят по вольт-амперной кривой (полярограмме), получаемой при электролизе исследуемого раствора в особом приборе – полярографе.
Один из электродов полярографа должен иметь очень маленькую поверхность. Обычно в качестве такого электрода служат капли ртути, вытекающие из капиллярного отверстия. Этот электрод является отрицательно заряженным катодом. Положительно заряженный анод представляет собой неподвижный электрод с большой площадью поверхности. Обычно это слой ртути на дне сосуда, в котором находится анализируемый раствор.
В ходе анализа постепенно увеличивают внешнюю разность потенциалов. Сначала ток через раствор почти не протекает, до тех пор, пока не будет достигнут потенциал разложения электролита.
При прохождении через раствор постоянного тока основное изменение концентрации ионов, по сравнению с их концентрацией в толще раствора, происходит у электрода с меньшей поверхностью (капельный электрод). Это обусловлено большей силой тока, приходящейся на единицу поверхности электрода, то есть более высокой плотностью тока. На капельном электроде возникает значительная концентрационная поляризация.
По мере повышения разности потенциалов между электродами увеличивается сила тока, протекающего через раствор, и плотность тока на капельном электроде. При этом скорость обеднения раствора восстанавливающимися ионами у поверхности капельного электрода также возрастает. В конечном итоге наступит такой момент, когда все движущиеся к катоду ионы приэлектродного слоя успевают разрядиться. Поскольку приэлектродный слой пополняется ионами медленнее, чем протекает процесс разрядки ионов на поверхности электрода, то дальнейшее повышение разности потенциалов не вызовет возрастания силы тока, и на полярограмме появится горизонтальный участок.
На рисунке 17.2 приведен общий вид полярограммы раствора, содержащего несколько катионов. Если в растворе присутствует один разряжающийся ион, то полярограмма имеет одну волну, при наличии нескольких ионов – несколько волн. По значению потенциала полуволны Е1/2 определяется вид ионов, а по величине предельного тока Iпр – их концентрация








 I, А
I, А
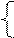 | |||
 | |||
(Iпр)3
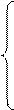 |
(Iпр)2
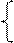 |
(Iпр)1
(Е1/2)1 (Е1/2)2 (Е1/2)3 Е, В
Рисунок 17.2 – Полярограмма раствора, содержащего несколько катионов
Кондуктометрия основана на измерении электропроводности разбавленных растворов электролитов, которая при прочих постоянных условиях опыта пропорциональна концентрации. Этим методом можно определять концентрацию электролита в разбавленном растворе, сравнивая полученное значение электропроводности с данными калибровочного графика, построенного для стандартных растворов (растворов с точно известной концентрацией).
При кондуктометрическом титровании фиксируют изменение электропроводности системы в ходе химической реакции. Точку эквивалентности определяют по резкому изменению электропроводности системы.
Остановимся теперь на спектральных методах анализа.
При взаимодействии анализируемого вещества с электромагнитными волнами может происходить поглощение, отражение или испускание излучения. Испускание лучей происходит также при нагреве вещества, воздействии радиоактивного излучения, потока электронов и других факторов. Эти явления используются в спектральных методах анализа вещества.
Методы, основанные на изучении спектров поглощения электромагнитного излучения, получили название абсорбционно-спектральных методов. Исследования проводят в инфракрасной, ультрафиолетовой, видимой областях спектра, а также в области радиоволн, чаще всего с длиной волны от5 до 0,6 м.
При взаимодействии любого типа излучения с веществом энергия поглощается порциями (квантами). Энергия кванта связана с частотой поглощаемого излучения соотношением
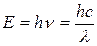 , (17.5)
, (17.5)
где h – постоянная Планка;
n – частота излучения;
c – скорость света в вакууме;
l – длина волны.
При взаимодействии инфракрасного (ИК) излучения с веществом происходит возбуждение колебаний валентно связанных атомов друг относительно друга. Эти колебания можно разделить на валентные (попеременное растягивание и укорочение ковалентной связи) и деформационные (изменение валентного угла между двумя связями одного атома).
Если через вещество пропускать ИК-излучение, медленно изменяя его частоту, то поглощение излучения будет наблюдаться только при тех частотах, которые совпадают с собственными частотами колебаний атомов в молекуле. Измеряя изменение интенсивности проходящего через вещество потока излучения, можно получить ИК-спектр поглощения данного вещества, отражающий зависимость интенсивности поглощения от частоты (точнее волнового числа 1/l, измеряемого в см-1).
ИК-излучение поглощают только те молекулы, в которых имеются полярные связи. Каждый тип связи поглощает излучение, характерной для него частоты. Одни и те же функциональные группы, входящие в состав молекул различных веществ, имеют практически одни и те же характеристические полосы поглощения (интервалы частот поглощаемого излучения). Отождествить полосы поглощения с определенными функциональными группами можно при помощи специальных справочных таблиц.
Таким образом, с помощью ИК-спектроскопии можно исследовать строение соединений, идентифицировать вещества, изучать внутримолекулярные и межмолекулярные взаимодействия, контролировать ход реакций.
Спектры поглощение в ультрафиолетовой (УФ) и видимой областях спектра обусловлены возбуждением молекул вещества и последующими переходами валентных электронов со связывающих и несвязывающих молекулярных орбиталей на более высокие по энергии разрыхляющие орбитали.
УФ-спектроскопия и спектроскопия в видимой области используются для установления строения молекул и наличия в них определенных хромофорных группировок, имеющих, как правило, кратные связи. Кроме того, при помощи этих методов возможно количественное определение вещества. Оно основано на законе Бугера – Ламберта – Бера, выражаемого соотношением
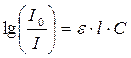 , (17.6)
, (17.6)
где 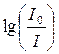 – оптическая плотность анализируемого раствора;
– оптическая плотность анализируемого раствора;
I0 – интенсивность потока света, падающего на раствор;
I – интенсивность потока света, прошедшего через раствор;
e – молярный коэффициент поглощения света;
l – толщина светопоглощающего слоя раствора;
С – молярная концентрация анализируемого раствора.
Измерив изменение интенсивности потока света, можно по формуле (17.6) определить концентрацию раствора анализируемого вещества. Измерения ведут при помощи спектрофотометров и фотоколориметров.
Спектрофотометрия основана на измерении светопоглощения при строго определенной длине волны облучающего света, которая соответствует максимуму кривой поглощения исследуемого вещества.
Фотоколориметрия основана на использовании видимого света. Анализируемые растворы при этом должны быть окрашены. Интенсивность их окраски связана с оптической плотностью раствора. Оптическую плотность анализируемого раствора сравнивают с данными калибровочного графика зависимости оптической плотности от концентрации, построенного для стандартных растворов, и таким образом определяют концентрацию раствора анализируемого вещества.
Суть нефелометрического метода анализа заключается в том, что о содержании определяемого вещества судят по интенсивности помутнения исследуемого раствора при действии определенного реагента. Этот метод основан на отражении света твердыми частицами, взвешенными в растворе.
В радиоволновой области электромагнитных волн также можно получить ценную информацию о структуре соединений. Одним из основных методов радиоспектроскопии является метод ядерного магнитного резонанса (ЯМР).
Ядра некоторых изотопов, например, 1H; 13C; 15N; 17O и другие, обладают ядерным спином. В отсутствии внешнего магнитного поля спины ориентированы беспорядочно, но в магнитном поле возможны лишь две ориентации спина – в направлении этого поля и против него. Состояние, при котором спиновый магнитный момент ориентирован по полю, имеет несколько меньшую энергию, чем состояние, при котором спиновый магнитный момент ориентирован против внешнего магнитного поля. Под воздействием радиочастотного электромагнитного излучения ядра атомов переходят с более низкого энергетического уровня на более высокий, поглощая порции энергии, соответствующие строго определенным частотам поглощаемого излучения.
Постепенно изменяя частоту излучения, можно наблюдать появление пиков поглощения, соответствующих резонансным сигналам всех ядер данного типа (например, протонов), содержащихся в молекуле. Так получают спектр ЯМР.
Для установления природы и строения радикалов, частиц с неспаренными электронами, применяется спектроскопия электронного парамагнитного резонанса (ЭПР). Физическая сущность этого метода аналогична сущности ЯМР. Разница состоит в том, что для переориентации спина неспаренного электрона нужна более значительная энергия, чем для переориентации спина ядра.
Методы эмиссионного спектрального анализа базируются на изучении спектров излучения. Вещество нагревается до высоких температур (2000 – 15000 0С). При этом оно испаряется, диссоциируя на атомы или ионы. Возбужденные атомы испускают электромагнитные волны. Проходя через спектрограф, излучение разлагается на присутствующие в нем компоненты в виде спектра цветных линий. Спектры испускания весьма специфичны, это своего рода «паспорт» элемента. Сравнивая спектр со справочными данными, определяют вид элемента, а по интенсивности спектральных линий – его содержание.
Существует группа люминесцентных методов анализа, основанных на свечении анализируемого вещества под воздействием ультрафиолетовых, рентгеновских, радиоактивных и других излучений и измерении интенсивности излучаемого веществами света.
Радиометрические методы анализа основаны на измерении излучений, испускаемых радиоактивными элементами.
Коснемся очень кратко сути хроматографических методов анализа. Хроматографические методы разделения и идентификации веществ основаны на том, что вещества помещают в систему, которая содержит два компонента, представляющие собой подвижную и неподвижную фазы. Молекулы веществ по-разному распределяются между этими фазами. Перемещающаяся подвижная фаза увлекает за собой вещество, что обусловливает его миграцию, в ходе которой вещество непрерывно перераспределяется между двумя фазами. Скорость движения определяемого вещества, из-за сродства к неподвижной фазе, всегда меньше скорости движения подвижной фазы. Скорость миграции вещества зависит от соотношения степеней его сродства к подвижной и неподвижной фазам. Если эти соотношения для компонентов анализируемой смеси различны, то они мигрируют с различными скоростями, и их удается отделить друг от друга, после того как они пройдут путь, достаточный для разделения.
Для осуществления хроматографического разделения требуется система, обеспечивающая направленное перемещение, например, трубка с сорбентом (колонка), полоска фильтровальной бумаги, тонкий слой сорбента на пластинке.
Неподвижная фаза может быть твердой, жидкой или представлять собой нелетучую жидкость, нанесенную на твердое тело. Подвижная фаза может быть жидкой или газообразной. Она обычно пропускается через неподвижную фазу или течет по ней.
Природа сродства вещества и неподвижной фазы может быть различной. В адсорбционной хроматографии это обратимая сорбция, в распределительной хроматографии – предпочтительная растворимость, в ионообменной хроматографии – электростатическое взаимодействие, в проникающей хроматографии – пребывание молекул внутри гранул пористой структуры, в аффинной хроматографии – специфическое биологическое взаимодействие.
Завершим рассмотрение инструментальных методов анализа масс-спектрометрическими методами. В масс-спектрометрическом анализе молекулы вещества, находящиеся в парообразном состоянии в глубоком вакууме, подвергаются ионизации, чаще всего пучком электронов с высокими энергиями (25 – 70 эВ).Молекулы в этих условиях теряют один электрон с наиболее высоких по энергии молекулярных орбиталей и превращаются в однозарядные молекулярные катионы, которые затем распадаются на осколки – ионы, свободные радикалы или молекулы. В масс-спектроскопии регистрируются только катионы. Они ускоряются в электрическом поле и попадают в магнитное поле, направленное перпендикулярно потоку катионов. В магнитном поле катионы с одинаковым отношением массы к заряду (m/e) разделяются на отдельные пучки, принимаются детекторами, а регистрирующие устройства тем или иным способом позволяют оценить относительное количество ионов определенной массы.
Совокупность относительных значений количества заряженных частиц для каждого из имеющихся значений m/e представляет масс-спектр данного вещества. Масс-спектры индивидуальных соединений обладают высокой специфичностью. Это позволяет идентифицировать исследуемое соединение по его масс-спектру. Масс-спектроскопия дает возможность установить молекулярную массу соединений, детали их строения, элементарный и изотопный состав.