КОЛИЧЕСТВЕННАЯ ТЕОРИЯ ОРГАНИЧЕСКИХ РЕАКЦИЙ
Часть I. Механизмы реакций органических соединений
Текст лекций

Санкт-Петербург
УДК 547.1.544 + 547.127
Целинский И.В. Количественная теория органических реакций. Часть I. Механизмы реакций органических соединений: текст лекций/ И.В.Целинский, С.Ф.Мельникова - СПб.: СПбГТИ (ТУ), 2010- 51с.
В данном учебном пособии излагаются основные принципы классификации механизмов органических реакций, рассматриваются главные типы гомолитических и гетеролитических процессов, синхронных реакций и перегруппировок, их механизмы, а также стереохимические аспекты реакций замещения и элиминирования.
Предназначено для студентов 3-курса по специальности 24.07.01-«Химическая тех- нология органических соединений азота» и 24.04.02-«Химическая технология синтетических биологически активных веществ», а также для аспирантов и научных сотрудников, специализирующихся в области физической органической химии и химической технологии органических веществ.
Ил.2 табл.
Рецезенты
1. СПб Госуниверситет кино и телевидения,
Л.Л.Кузнецов, д-р. хим. наук, профессор кафедры
общей, органической и физической химии
2. Б.М.Ласкин, д-р техн.наук, профессор, начальник
научно-исследовательского комплекса «Экология
промышленная безопасность «РНЦ «Прикладная химия»
Утверждено на заседании учебно-методической комиссии факультета тонкого органического и микробиологического синтеза 2010 г.
Рекомендовано к изданию РИС’о СПбГТИ (ТУ)
ВВЕДЕНИЕ
Количественная теория органических реакций опирается, прежде всего, на знание механизмов химических реакций и факторов, определяющих реакционную способность, прежде всего структурных (электронные, стереоэлектронные и стерические эффекты) и эффектов среды (клеточный эффект, специфическая и неспецифическая сольватация). Разобраться во множестве химических превращений органических веществ помогает классификация их механизмов, которая проводится по двум основным принципам: по характеру разрыва и образования химических связей (гомолитические, Гетеролитические реакции, процессы одноэлектронного переноса) и по типу превращений субстрата (реакции присоединения, замещения, элиминирования, фрагментации). Отдельный тип органических реакций представляют синхронные, перициклические и электроциклические реакции, а также сигматропные перегруппировки.
Органические реакции различаются также по типу активации. Большинство некаталитических реакций, скорость которых увеличивается с температурой, относятся к числу термически активируемых*. В последнее время широкое распространение получили реакции, активируемые действием микроволнового облучения, в основе которого также лежит термическая активация. Отдельную группу представляют т.н. сонохимические реакции, активируемые ультразвуком, однако этот метод активации используется гораздо реже, чем термическая активация.
Принципиально отличными от приведенных выше являются фотохимические реакции, активируемые ультрафиолетовым облучением, и радиохимические реакции, протекающие под действием ионизирующего излучения. В таких реакциях органические молекулы, поглощая квант электромагнитного излучения или сталкиваясь с элементарной частицей, переходят в электронно-возбужденное состояние или распадаются на фрагменты, реакционная способность которых принципиально отличается от молекул в основном электронном состоянии. Такие реакции лишь вскользь упоминаются в данном учебном пособии.
*Каталитическая активация органических реакций подробно рассмотрена в учебном пособии «Кислотно-основные свойства органических соединений азота» авторов И.В.Целинский и И.В.Шугалей, СПб; СПбГТИ(ТУ), 2006.-57 с.
Классификация механизмов органических реакций по характеру разрыва и образования химических связей.
По данному принципу реакции подразделяются на гомолитические (или свободнорадикальные), гетеролитические, или ионные, и реакции одноэлектронного переноса (или окислительно-восстановительные).
1.1 Гомолитические реакции
Элементарной стадией свободно-радикальных (гомолитических) реакций является гомолитический разрыв ковалентной связи, так что каждый из двух образовавшихся фрагментов несет неспаренный электрон, либо обратный процесс рекомбинации двух свободных радикалов, либо взаимодействие свободного радикала с нейтральной молекулой или ионом. В соответствии с этим различают следующие типы гомолитических реакций:
1.1.1 Гомолитическая диссоциация (распад молекулы на радикалы)
Молекулы органических соединений, имеющие в своем составе слабые ковалентные сигма-связи (с энергией диссоциации до 150-170 кДж/моль), такие как связи N-N, N-O, O-O, N-Cl (Br) и другие, при нагревании (обозначаемом знаком Δ) диссоциируют с образованием свободных радикалов, например, перекись бензоила образует два бензоильных радикала

а диметилнитрамин – диоксид азота и диметиламинильный радикал

Возможен также согласованный распад двух сигма-связей, как в случае термической диссоциации инициатора свободно-радикальной полимеризации – азобис-изо-бутиронитрила

1.1.2 Соединение свободных радикалов друг с другом (рекомбинация радикалов)
Это процесс, обратный предыдущему - например, одна из стадий радикального нитрования алканов

1.1.3  Гомолитическое замещение имеет место, когда один свободный радикал вытесняет другой из нейтральной молекулы, например, при образовании арилалкилсульфидов из дисульфидов:
Гомолитическое замещение имеет место, когда один свободный радикал вытесняет другой из нейтральной молекулы, например, при образовании арилалкилсульфидов из дисульфидов:
 или путем вытеснения трифторметильным радикалом метильного из тетраметилсилана:
или путем вытеснения трифторметильным радикалом метильного из тетраметилсилана:
 1.1.4 Гомолитическое присоединение свободных радикалов к нейтральным молекулам, чаще всего к олефинам:
1.1.4 Гомолитическое присоединение свободных радикалов к нейтральным молекулам, чаще всего к олефинам:
Такие реакции представляют собой, в частности, стадии инициирования и продолжения цепи свободнорадикальной полимеризации алкенов.
 1.1.5 Гомолитическое расщепление радикалов приводит к образованию нейтральной молекулы и нового, более низкомолекулярного радикала (или атома). Например, этоксильный радикал может распадаться по двум направлениям:
1.1.5 Гомолитическое расщепление радикалов приводит к образованию нейтральной молекулы и нового, более низкомолекулярного радикала (или атома). Например, этоксильный радикал может распадаться по двум направлениям:
 Бензоильный радикал распадается с выделением диоксида углерода:
Бензоильный радикал распадается с выделением диоксида углерода:
 1.1.6 Диспропорционирование радикалов. Передача атома (чаще всего водорода) или группы от одного свободного радикала к другому приводит к образованию двух нейтральных молекул, например:
1.1.6 Диспропорционирование радикалов. Передача атома (чаще всего водорода) или группы от одного свободного радикала к другому приводит к образованию двух нейтральных молекул, например:
 1.1.7 Изомеризация радикалов, приводящая к образованию более стабильного свободного радикала:
1.1.7 Изомеризация радикалов, приводящая к образованию более стабильного свободного радикала:
Свободный радикал (А), обладая незаполненной электронной оболочкой и проявляя в силу этого электрофильный* характер, стабилизируется за счет трех электронодонорных метильных групп при образовании трет.-бутильного радикала (В). Это является движущей силой изомеризации
1.2 Гетеролитические, или ионные реакции.
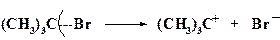 Гетеролитические процессы протекают с участием ионов, т.е. частиц, несущих электрический заряд, образующихся при гетеролизе ковалентной связи. При этом пара электронов сигма-связи переходит к аниону (отрицательно заряженной частице), а катион, т.е. положительно заряженная частица в случае элементов II периода останется с незаполненной 6-электронной оболочкой. Например, трет.-бромистый бутил спонтанно диссоциирует с образованием бромид-аниона и трет.-бутильного катиона.
Гетеролитические процессы протекают с участием ионов, т.е. частиц, несущих электрический заряд, образующихся при гетеролизе ковалентной связи. При этом пара электронов сигма-связи переходит к аниону (отрицательно заряженной частице), а катион, т.е. положительно заряженная частица в случае элементов II периода останется с незаполненной 6-электронной оболочкой. Например, трет.-бромистый бутил спонтанно диссоциирует с образованием бромид-аниона и трет.-бутильного катиона.
 Ионы, обладая электрическим зарядом, являются активными промежуточными частицами во многих химических реакциях. Ионы могут образовываться в процессе кислотно-основных превращений (ионизация кислот, протонирование оснований и т.д.):
Ионы, обладая электрическим зарядом, являются активными промежуточными частицами во многих химических реакциях. Ионы могут образовываться в процессе кислотно-основных превращений (ионизация кислот, протонирование оснований и т.д.):

1.2.1 Карбокатионы
Весьма распространенным процессом в органической химии является образование карбокатионов (или карбениевых ионов) с положительным зарядом, локализованным на атоме углерода. Карбокатионы в большинстве случаев являются весьма активными промежуточными продуктами (интермедиатами) и легко вступают в дальнейшие гетеролитические процессы.
Образование карбокатионов возможно в результате спонтанной диссоциации алкил-
___________________________________________________________________________
*Понятие электрофильности и нуклеофильности см. далее в разделе “Гетеролические и ионные реакции” (стр.14)
 и аралкилгалогенидов, протекающей в среде полярных (т.н. ионизирующих) растворителей
и аралкилгалогенидов, протекающей в среде полярных (т.н. ионизирующих) растворителей
Такие реакции называются процессами сольволиза. Карбокатионы, будучи электроно-дефицитными частицами, стабилизируются за счет положительного индукционного (передаваемого по простым сигма-связям) или мезомерного (передаваемого по системе сопряженных кратных связей) эффектов электронодонорных групп

Заметим, что при образовании карбокатионов меняется гибридизация карбениевого центра от sp3 (тетраэдрической) в исходном соединений до sp2 (плоской тригональной) в катионе, что способствует стабилизации этих ионов.
Карбокатионы могут образовываться также под действием на исходные спирты (или их производные) кислот Бренстеда (т.е. протонных кислот):

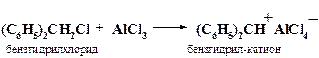 либо действием на алкил(аралкил)галогениды кислот Льюиса (т.е. апротонных кислот):
либо действием на алкил(аралкил)галогениды кислот Льюиса (т.е. апротонных кислот):
1.2.2 Катионы с зарядом на гетероатоме.
Важную роль в органической химии играют катионы, положительно заряженным центром которых являются атомы азота, кислорода, серы и галогенов. Аммониевые катионы образуются при протонировании аминов (иминов) и различных азотистых оснований. Наличие неподеленной пары электронов (НПЭ) на атоме азота обуславливает достаточно высокую основность аминов*

 Ароматические амины и азотистые гетероциклы (пиррол, пиразол, имидазол, триазолы, тетразол, пиридин, пиразин, пиримидин и т.д.), хотя и являются более слабыми основаниями, чем аммиак и алкиламины, также способны протонироваться под действием сильных кислот:
Ароматические амины и азотистые гетероциклы (пиррол, пиразол, имидазол, триазолы, тетразол, пиридин, пиразин, пиримидин и т.д.), хотя и являются более слабыми основаниями, чем аммиак и алкиламины, также способны протонироваться под действием сильных кислот:

Ионы иммония образуются при протонировании алкил(арил, аралкил, гетерил) иминов:
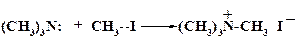 Четвертичные аммониевые катионы образуются при алкилировании триалкиламинов галогеналканами, например,
Четвертичные аммониевые катионы образуются при алкилировании триалкиламинов галогеналканами, например,
 Ионы гидроксония и алкилгидроксония (H3O+ и ROH2+) генерируются при протонировании воды и спиртов:
Ионы гидроксония и алкилгидроксония (H3O+ и ROH2+) генерируются при протонировании воды и спиртов:
а ионы триалкилоксония - при алкилировании гидроксониевых солей диазометаном или алкилгалогенидами в присутствии кислот Льюиса

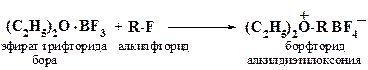
__________________________________________________________________________________
*Подробнее эти вопросы рассмотрены в учебном пособии «Кислотно-основные свойства органических соединений азота» авторов И.В.Целинский и И.В.Шугалей; СПб, СПбГТИ (ТУ), 2006.-57с
Ион гидросульфония образуется при протонировании серного ангидрида:

 Ионы галогенония (Cl+, Br+, I+) возникают при гетеролизе связи Hgl-Hgl в молекулах галогенов, протекающем обычно под действием кислот Бренстеда или Льюиса:
Ионы галогенония (Cl+, Br+, I+) возникают при гетеролизе связи Hgl-Hgl в молекулах галогенов, протекающем обычно под действием кислот Бренстеда или Льюиса:

Ионы нитрозония (NO+) и нитрония (NO2+) образуются в результате дегидратации протонированных под действием сильных кислот, соответственно, азотистой и азотной кислот
а ион диазония (R-N+≡N, обычно арил- или гетарилдиазония) генерируются путем дегидратации диазогидратов - таутомерной формы нитрозоаминов:

1.2.3 Карбанионы.
Важными промежуточными частицами в органических реакциях являются карбанионы, R3C-, т.е. частицы с отрицательным зарядом, образовавшиеся после отрыва протона основанием от исходной т.н. CH-кислоты.


 Простые углеводороды являются нейтральными соединениями и практически не проявляют кислотных свойств даже при действии очень сильных оснований, за исключением ацетилена и его производных (рКа 23-25), циклопентадиена
Простые углеводороды являются нейтральными соединениями и практически не проявляют кислотных свойств даже при действии очень сильных оснований, за исключением ацетилена и его производных (рКа 23-25), циклопентадиена
индена флуорена
и их аналогов.
Кислотность ацетиленов обусловлена повышенной электроотрицательностью sp -гибридизированного атома углерода, т.е. его способностью удерживать отрицательный заряд аниона. Такой же эффект при наличии более электроотрицательного атома азота у тройной связи в молекуле синильной кислоты (H-C≡N) приводит к существенному усилению кислотности (рКа 9.3).
Кислотность циклопентадиена, индена, флуорена и их производных приписывается эффекту ароматизации соответствующих карбанионов. Согласно правилу Хюккеля, ароматическими считаются циклические соединения, содержащие систему сопряженных кратных связей с (4n+2) π–электронами, где n=0,1,2,3 и т.д. Классическим ароматическим соединением является бензол, в молекуле которого имеется 6π-электронов на трех сопряженных кратных связях. При этом все шесть связей С-С молекулы бензола одинаковы, что свидетельствует о делокализации π-электронов в цикле. Такая делокализация электронов стабилизирует молекулу бензола примерно на 150 кДж/моль.
Анионы циклопентадиена, индена и флуорена являются изоэлектронными аналогами, соответственно, бензола (6π-электронов), нафталина (10π-электронов) и антрацена (14π-электронов) и также стабилизированы сопряжением (резонансом).
Карбанионы могут стабилизироваться также за счет эффекта полярного сопряжения при наличии у реакционного центра таких –М (обладающих отрицательным мезомерным эффектом) заместителей, как нитро-, циано-, сульфонильная, карбонильная, карбалкоксильная и другие группы, которые называются ацидифицирующими (т.е. придающими молекуле кислотные свойства) заместителями:

(Обоюдоострыми стрелками на этой схеме обозначаются т.н. крайние, или предельные резонансные (мезомерные) структуры, описывающие возможное распределение отрицательного заряда аниона).
Наиболее мощным –М - эффектом среди этих заместителей обладает нитрогруппа. Введение второго и третьего –М - заместителя существенно усиливает ацидифицирующий эффект за счет расширения области делокализации отрицательного заряда карбаниона:

Значения рКа ди- и тризамещенных –М-группами производных метана приведены ниже: CH2(NO2)2 3.6; CH(NO2)3 0.1; (CH3CO)2CH2 9.0; (CH3CO)3CH 5.8; CH2(CN)2 11.2; CH(CN)3 -5.1; CH(NO2)2CN -6.2.
Менее эффективно ацидифицируют углеводороды электроноакцепторные заместители, такие как галогены, действующие по механизму индукционного эффекта, т.е. путем поляризации простых σ -связей, например:
Cl3CH, pKa 15.5; C6F5H, pKa 25.8; F3CH, pKa 26.5; (CF3)3CH, pKa 11.0.
1.2.4 Реакции ионов.
1.2.4.1 Перегруппировка ионов с образованием более стабильных изомеров.
Наиболее хорошо изучены часто встречающиеся и имеющие важное практическое
 применение перегруппировки карбокатионов. Обычно они катализируются кислотами Бренстеда или Льюиса и включают первичный акт присоединения протона к молекуле алкана с образованием карбониевого катиона с пятикоординационным атомом углерода:
применение перегруппировки карбокатионов. Обычно они катализируются кислотами Бренстеда или Льюиса и включают первичный акт присоединения протона к молекуле алкана с образованием карбониевого катиона с пятикоординационным атомом углерода:
 На следующей стадии при отщеплении молекулы водорода образуется трехкоординационный катион (ион карбения), который перегруппировывается в более стабильный катион:
На следующей стадии при отщеплении молекулы водорода образуется трехкоординационный катион (ион карбения), который перегруппировывается в более стабильный катион:
Третичный бутильный катион более стабилен, чем вторичный бутильный, за счет электронодонорного эффекта метильных групп, частично нейтрализующих положительный заряд катиона. Такие изменения углеродного скелета с участием карбокатиона называются перегруппировками Вагнера-Меервейна.
Возможны также перегруппировки без изменения скелета, например,
превращение первичного пропильного катиона в более стабильный вторичный путем т.н. 1,2-гидридного сдвига, т.е. перемещения атома водорода с двумя электронами

Перегруппировки с участием карбанионов встречаются значительно реже в связи с необходимостью делокализации четырех электронов в переходном состоянии (ПС) вместо двух в случае карбокатионов:

Известны 1,2-сдвиги арильных групп, например, в реакции 1,1,1-трифенил-2-хлорэтана с натрием, протекающие с промежуточным образованием карбаниона:
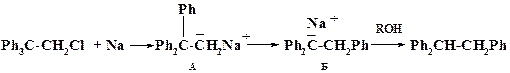
Карбанион Б стабилизирован делокализацией отрицательного заряда на двух фенильных группах, что обеспечивает движущую силу процесса.
В перегруппировке Стивенса происходит 1,2-сдвиг алкильной группы от атомов азота или серы к атому углерода карбаниона:

1.2.4.2 Распад иона на более простой ион и молекулу.
Одним из вариантов стабилизации карбокатионов является отщепление протона с образованием алкенов:

Алкокси-анионы, например, анионы β, β-динитроспиртов или анион ацетонциангидрина, легко расщепляются в щелочной среде на более термодинамически стабильные анионы 1,1-динитроалканов или цианид-анион с выделением карбонильного соединения (реакция ретро-Анри)

1.2.4.3 Присоединение ионов к нейтральным молекулам.
 Такие реакции характерны как для катионов (электрофильное присоединение), так и для анионов (нуклеофильное присоединение) и протекают с участием непредельных соединений, содержащих связи С=С, С=N, С=O и т.д., и соединений с малыми карбо- и гетероциклами (циклопропан, азиридин, оксиран, оксетан). Например, галогенирование алкенов:
Такие реакции характерны как для катионов (электрофильное присоединение), так и для анионов (нуклеофильное присоединение) и протекают с участием непредельных соединений, содержащих связи С=С, С=N, С=O и т.д., и соединений с малыми карбо- и гетероциклами (циклопропан, азиридин, оксиран, оксетан). Например, галогенирование алкенов:
Кислотно-катализируемая гидратация алкенов:

Присоединение карбокатионов к алкенам:

:
 Реакция Михаэля:
Реакция Михаэля:
Электрофильное присоединение к кратным связям
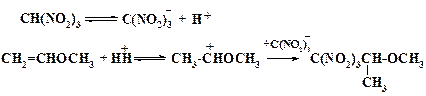
Реакции раскрытия циклов


1.2.4.4 Замещение ионами других ионов в нейтральных молекулах.
Это широко распространенный тип реакций в органической химии. Подобные процессы протекают в рядах насыщенных алифатических, непредельных, ароматических и гетероциклических соединений с участием катионов, анионов и нейтральных соединений, генерирующих ионы при действии катализаторов либо спонтанно.
 Например, нуклеофильное замещение у насыщенного атома углерода:
Например, нуклеофильное замещение у насыщенного атома углерода:
 Электрофильное замещение в ароматических соединениях:
Электрофильное замещение в ароматических соединениях:
Нуклеофильное замещение в ароматическом ряду

1.2.4.5 Рекомбинация с ионом противоположного знака.
Сюда относятся процессы, обратные реакциям гетеролиза ковалентной связи, например, нейтрализация Бренстедовских кислот основаниями:

а также процесс, обратный сольволизу алкилгалогенидов:

Взаимодействие неорганических катионов с анионами приводит к образованию нейтральных молекул:

1.2.5 Нуклеофильные и электрофильные реагенты и реакции.
Гетеролитические реакции и реагенты подразделяются на нуклеофильные и электрофильные. Химические связи в этих реакциях образуются таким образом, что одна частица является носителем пары электронов, а другая имеет свободную или освобождающуюся в ходе реакции орбиталь для образования ковалентной связи. Носитель пары электронов, отдающий ее для образования связи с субстратом, называется нуклеофильным реагентом, или нуклеофилом, а частица, предоставляющая свободную орбиталь для образования ковалентной связи – электрофильным реагентом, или электрофилом.
 Например, в реакции замещения
Например, в реакции замещения
этоксильный анион (реагент) является нуклеофилом, а метилиодид (субстрат) – электрофилом.
В реакции ароматического нитрования нитроний-ион (реагент) является электрофилом, а бензол (субстрат)- нуклеофилом.

Нуклеофильными реагентами могут быть:
- Анионы: Н ¯ (гидрид), ОН ¯ (гидроксид), RO ¯ (алкоксид), ArO ¯ (арилоксид или фенолят), HOO ¯ (гидропероксид), RCOO ¯ (ацилоксид или карбоксилат), HS ¯ (гидросульфид), RS ¯ (алкилтиолат), RC(O)S ¯ (тиокарбоксилат), ArS ¯ (тиофенолят), NH2 ¯ (амид), R2N ¯ (диалкиламид), N3 ¯ (азид), CN ¯ (цианид), NCO ¯ (цианат), SCN ¯ (тиоцианат), F ¯ (фторид), Cl ¯ (хлорид), I ¯ (иодид), NO2 ¯ (нитрит), NO3 ¯ (нитрат) и другие, в том числе карбанионы: R2C¯NO2 (нитронат), CH3C(O)CH2¯(ацетонат), ¯CH2C≡N (анион ацетонитрила), (CH3CO)2CH¯(ацетилацетонат), Cl3C¯(трихлорметид), Ar2CH¯ (бензгидрильный анион),  RC(NO2)2¯(гем-динитроалкильный анион или 1,1-динитрокарбанион), Ar3C¯ (триарилметильный анион), RC≡C¯ (ацетиленид-анион), C¯≡N (цианид-анион),
RC(NO2)2¯(гем-динитроалкильный анион или 1,1-динитрокарбанион), Ar3C¯ (триарилметильный анион), RC≡C¯ (ацетиленид-анион), C¯≡N (цианид-анион),
(циклопентадиенил-анион),¯CH(CN)2 (анион малононитрила), (C2H5OCO)2CH¯ (диэтилмалонат-ион) и т.д.
Нейтральные молекулы, имеющие атомы с неподеленными парами электронов: вода (H2O), спирты (ROH), фенолы (ArOH), аммиак (NH3), амины (R3N), гидразины (RNHNH2), гидроксиламины (RNHOH), фосфин (PH3).
Положительно заряженные (или нейтральные) реагенты, принимающие в ходе реакции электронную пару для образования ковалентной связи с субстратом, как сказано выше, называются электрофилами.
К ним относятся:
- катионы H+ (гидроний), Cl+ (хлороний), Br+ (бромоний), NO+ (нитрозоний), NO2+ (нитроний), ArN+≡N (арилдиазоний), HSO3+ (гидросульфоний), катионы металлов (Ag+,Hg2+ и др.),
- карбокатионы R3C+(триалкилкарбениевый ион* или триалкилкарбокатион), Ar3C+ (триарилкарбениевый ион), C6H5CH2+ (бензил-катион), CH2=CHCH2+ (аллил-катион),
+CH2OH (гидроксиметил-катион), +CH2Cl (хлорметил-катион), R2N-CH2+ (карбений-иммониевый ион), R-C+ =O (ацилий-катион).
- кислоты Льюиса: BF3, AlCl3, ZnCl2, SnCl4, TiCl4, SbF5 и т.д.
 - нейтральные молекулы, образующие катионы при гетеролизе, например,
- нейтральные молекулы, образующие катионы при гетеролизе, например,

- молекулы с кратными полярными связями
способные образовывать новую связь за счет пары электронов субстрата.
______________________________________________________________________
*Логичнее было бы назвать этот ион и аналогичные катионы карбониевыми ионами (подобно аммонииевым, оксониевым, сульфониевым ионам), однако это название закреплено за пятикоординированным углеродным катионом (например, CH5+-ион метония), в отличие от трехкоординированных карбениевых ионов.
 Разделение гетеролитических реакций на нуклеофильные и электрофильные в значительной мере условно, т.к. в бимолекулярной гетеролитической реакции участвуют как нуклеофильный, так и электрофильный компоненты. Например, в рассмотренной выше реакции нитрования бензола NO2+ является электрофилом, а C6H6-нуклеофилом.
Разделение гетеролитических реакций на нуклеофильные и электрофильные в значительной мере условно, т.к. в бимолекулярной гетеролитической реакции участвуют как нуклеофильный, так и электрофильный компоненты. Например, в рассмотренной выше реакции нитрования бензола NO2+ является электрофилом, а C6H6-нуклеофилом.
Однако в синтетической химии реагирующие вещества условно делят на реагенты и субстраты, причем субстратами чаще всего считается нейтральная молекула или частица с большей молекулярной массой (или большим количеством атомов). Реагенты часто являются неорганическими соединениями, поэтому в приведенной выше реакции NO2+ считается реагентом, а C6H6 –субстратом, и реакция считается процессом электрофильного замещения.
В нуклеофильной же реакции субстрат подвергается действию нуклеофильного реагента.