Гетеролитические реакции подразделяются на реакции присоединения (индекс Ad-от английского Addition), замещения (индекс S- от английского Substitution), элиминирования (индекс E- от английского Elimination) и фрагментации (индекс F- от английского Fragmentation).
Для более точной индексации механизма к основному символу добавляется подстрочный индекс N (нуклеофильное) или E (электрофильное), а также цифры 1 (мономолекулярное) или 2 (бимолекулярное).
2.1 Реакции замещения.
Замещение – это реакция, в ходе которой какой-либо атом или функциональная группа в органической молекуле замещается на другой атом(функциональную группу). В общем виде это можно записать следующим образом:

В приведенной выше реакции R-X - субстрат (где R-углеводородный радикал, а X - уходящая группа). Атакующий реагент Y называется вступающей группой.
Реакции замещения могут быть моно- и бимолекулярными (S1 и S2), нуклеофильными либо электрофильными (соответственно, SN и SE).
В реакции алифатического мономолекулярного замещения лимитирующей стадией является гетероциклическая диссоциация субстрата, и скорость всего процесса не зависит от концентрации атакующего реагента. При бимолекулярном замещении скорость реакции прямо пропорциональна концентрациям и реагента и субстрата.
Например, трет.-бромистый бутил подвергается спонтанной гетеролитической диссоциации, лимитирующей скорость процесса (SN1). Образующийся трет.-бутильный катион может либо отщеплять протон (реакция элиминирования), либо присоединяться к нуклеофилу Y- (например, цианид-ион CN-, азид-ион N3-) или же гидролизоваться в содержащем воду растворителе:
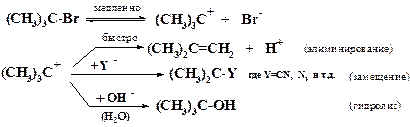
 Первичные и вторичные алкилгалогениды, в отличие от трет.-бутилгалогенидов, в обычных условиях не подвергаются мономолекулярной диссоциации, и для них характерно бимолекулярное нуклеофильное замещение (SN2), при котором разрыв связи R-X и образование новой связи Y-R происходят синхронно
Первичные и вторичные алкилгалогениды, в отличие от трет.-бутилгалогенидов, в обычных условиях не подвергаются мономолекулярной диссоциации, и для них характерно бимолекулярное нуклеофильное замещение (SN2), при котором разрыв связи R-X и образование новой связи Y-R происходят синхронно
(знаком ≠ в квадратных скобках обозначено переходное состояние реакции, обладающее максимумом потенциальной энергии).
Примером может служить реакция В.Мейера:

Реакции нуклеофильного ароматического замещения рассмотрены ниже (см.п.5.2).
Реакции электрофильного замещения (SE) характерны для металлорганических соединений, а также ароматических углеводородов. Они также могут быть как моно -, так и бимолекулярными:

Например, по такому механизму протекает замена атома ртути на ее изотоп (Hg*) в молекуле бензгидрилмеркурбромида:

а также магнийорганические синтезы с участием реактива Гриньяра:
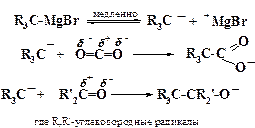
Бимолекулярное электрофильное замещение (SE2) характерно для ароматических углеводородов, например, нитрования бензола солями нитрония, т.е. замещение атома водорода на нитрогруппу
Отличие таких реакций от вышеприведенных процессов электрофильного замещения
в том, что вначале происходит образование новой связи C-NO2 в так называемом σ-комплексе, а затем от него отщепляется протон и образуется конечный продукт - нитробензол. Поэтому такие процессы обозначаются символом SEAr, т.е. электрофильное замещение в ароматических соединениях (аренах) (подробнее механизм рассмотрен ниже, п.6, стр.37).
2.2 Реакции присоединения.
Присоединение - это реакция, в ходе которой реагент присоединяется к субстратам, содержащим кратные связи (C=C, C=O, C=N,C≡N, C≡C и т.д.) или раскрывает малые
(3-и 4-членные) циклы. Это, например, бромирование алкенов:

Такие реакции относятся к процессам электрофильного присоединения (AdE), поскольку молекула брома поляризуется при образовании π-комплекса с олефином, проявляя свойства электрофильного реагента.
В лимитирующей стадии происходит образование карбениевого иона, который далее в быстрой стадии рекомбинирует с бромид-анионом, давая конечный продукт.
По аналогичному механизму протекает реакция присоединения к алкенам галогенводородов и сильных OH-, NH- и CH- кислот:

или
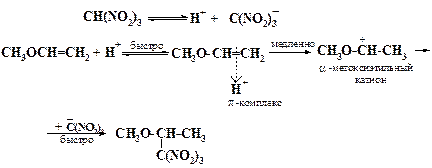
Лимитирующей стадией этих реакций является присоединение протона к атому углерода кратной связи (через промежуточное образование π-комплекса). Образующийся карбениевый ион быстро рекомбинирует с присутствующими в системе анионами (Cl-, C(NO2)3- и т.д.), образуя конечный продукт.
 Легче всего протекает присоединение к алкенам, содержащим донорные заместители
Легче всего протекает присоединение к алкенам, содержащим донорные заместители
, что характерно для реакций типа AdE.
Присоединение проходит по правилу Марковникова, т.е. протон присоединяется к наиболее гидрогенизованному атому углерода, давая более стабильный вторичный, а не первичный карбениевый ион.
 Реакции нуклеофильного присоединения (AdN) характерны для алкенов, содержащих в α-положении электроноакцепторные группы, проявляющие –М-эффект, такие как NO2, CN, COR, COOR, CONR2, SO2R и др. Под действием таких заместителей происходит поляризация кратной связи С=С, например,
Реакции нуклеофильного присоединения (AdN) характерны для алкенов, содержащих в α-положении электроноакцепторные группы, проявляющие –М-эффект, такие как NO2, CN, COR, COOR, CONR2, SO2R и др. Под действием таких заместителей происходит поляризация кратной связи С=С, например,
Это делает возможной атаку нуклеофилом (аминами, спиртами, анионами CH-, NH-, OH- и SH-кислот) концевой метиленовой группы, несущей дробный положительный заряд, с образованием в лимитирующей стадии промежуточного α-цианокарбаниона, который в последней быстрой стадии отрывает протон от сопряженной кислоты нуклеофила или растворителя, давая конечный продукт присоединения:
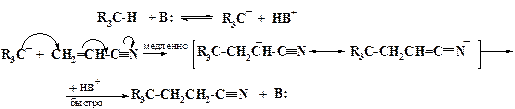
Эта реакция носит название реакции Михаэля. Продукт такой реакции обычно называют аддуктом.
2.3 Реакции элиминирования.
Элиминирование – это реакция, в ходе которой от субстрата отщепляется в виде двух фрагментов нейтральная молекула или заряженная частица (вода, спирт, амин, галогеноводород и т.д.).
В зависимости от взаимного расположения атомов углерода, от которых происходит отщепление этих фрагментов, различают α-, β -, γ - и т.д. элиминирование. Если оба отщепляющихся фрагмента связаны с одним и тем же атомом углерода, такой процесс называется α-элиминированием. В результате α-элиминирования образуется нейтральная высокореакционная частица с двухвалентным (двухкоординированным) атомом углерода, называемая карбеном:

Например, хлороформ (CHCl3) проявляет свойства слабой СН-кислоты (рКа 15.5) и под действием сильных оснований (В) отщепляет протон, а образующийся карбанион CCl3- подвергается α-элиминированию с выделением дихлоркарбена.

Если два отщепляющихся атома или группы связаны с соседними атомами углерода, то такой процесс называется β -элиминированием. В результате β -элиминирования образуются непредельные соединения (алкены):
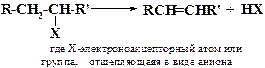
Известны также реакции γ-, δ - и т.д. элиминирования, при которых из линейных алифатических соединений образуются циклические продукты:

Для реакций β -элиминирования возможны три механизма, различающиеся последовательностью отщепления протона и уходящей группы X: E1, E1сВ и E2.
2.3.1 В механизме E1 (мономолекулярное элиминирование) лимитирующей (наиболее медленной) стадией является ионизация субстрата с образованием карбокатиона, который в последующей быстрой стадии отщепляет протон от β -углеродного атома, превращаясь в алкен:

Первая стадия этого процесса идентична лимитирующей стадии мономолекулярного нуклеофильного замещения (SN1). Таким образом, промежуточный карбокатион участвует в двух параллельных конкурирующих реакциях – присоединение нуклеофила и отщепление протона.
2.3.2. Мономолекулярное элиминирование в сопряженном основании субстрата (E1cB) также включает две стадии, но в обратной последовательности - отщепление протона предшествует отрыву уходящей группы
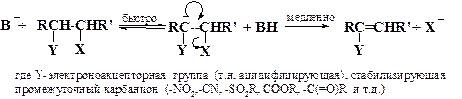
Поскольку интермедиатом здесь является карбанион, символ E1cB (cB-conjugate base) указывает, что уходящая группа Х отщепляется от сопряженного субстрату основания. Механизм E1cB описывается кинетическим уравнением общего второго порядка с первым частным порядком по субстрату и по основанию В - , однако реакция является мономолекулярной, поскольку в лимитирующей скорость стадии участвует одна частица (карбанион).
2.3.3 Бимолекулярное элиминирование (E2).
 В реакциях бимолекулярного элиминирования от молекулы субстрата одновременно отщепляются две группы - обычно это анионоидная уходящая группа Х (нуклеофуг) и протон, отщепляющийся от соседнего атома углерода:
В реакциях бимолекулярного элиминирования от молекулы субстрата одновременно отщепляются две группы - обычно это анионоидная уходящая группа Х (нуклеофуг) и протон, отщепляющийся от соседнего атома углерода:
Примером могут служить реакции дегидрогалогенирования галогеналканов под действием оснований:
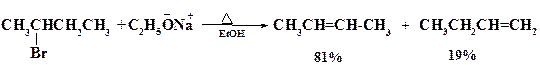
Процесс описывается кинетическим уравнением общего второго порядка, первого по субстрату и по основанию В-, аналогично реакциям SN2 -замещения. По сути, оба эти процесса конкурируют друг с другом, отличаясь лишь направлением атаки нуклеофильного реагента.
Таким образом, механизм Е2 является промежуточным между Е1 и Е1сВ, при этом переходное состояние может варьировать в зависимости от относительной степени расщепления связей C-H и C-X, которое не всегда происходит строго синхронно.
Наиболее важной особенностью Е2 -элиминирования является его стереоспецифичность. При синхронном элиминировании необходимо, чтобы оси p -орбиталей двух атомов углерода, от которых происходит элиминирование, были параллельны. Только в этом случае возможно образование π-связи. Это может быть достигнуто, когда группы Х и Н находятся в транс–положении друг к другу (заторможенная конформация А), либо, что менее вероятно, в цис-положении (заслоненная конформация В):
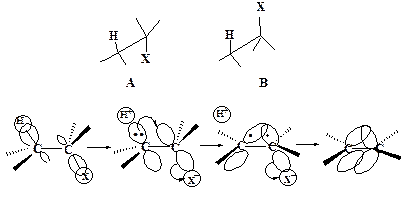
Конформация А носит название анти-перипланарной, а элиминирование групп Х и Н из нее называется анти-элиминированием.
Конформация В называется син-перипланарной и элиминирование групп Х и Н из нее носит название син-элиминирование. В общем случае анти-элиминирование предпочтительнее син-элиминирования по пространственным соображениям.
Как отмечено выше, бимолекулярное элиминирование (Е2) конкурирует с бимолекулярным нуклеофильным замещением (SN2). Сходство этих процессов в том, что скорость как Е2 -, так и SN2 реакций возрастает в ряду алкилгалогенидов
R-F < R-Cl < R-Br < R-I.
Различие же состоит в том, что сильные основания (t-BuO -, NH2 -) способствуют Е2 - реакции, а сильные нуклеофилы, но слабые и средние по силе основания (NH3, I -, NH2OH) способствуют реакции замещения SN2. Увеличение объема нуклеофила так-
же способствует процессу Е2 вследствие меньших стерических требований этой реакции. Более полярные растворители, наоборот, благоприятствуют процессам замещения (SN2), т.к. переходное состояние в этом случае более полярно и лучше сольватировано, что понижает его энергию. В общем энергия активации процессов Е2 выше, чем для SN2, поэтому повышение температуры способствует Е2 -элиминированию.
2.4 Мономолекулярная фрагментация (F1) представляет собой элиминирование катионных и анионных фрагментов субстрата с выделением нейтральной молекулы. Например, бензилазокситозилат (А) при нагревании в полярных растворителях распадается на монооксид диазота N2O, бензильный катион и тозилат (п-метилбензолсульфонат)-анион. Последние могут рекомбинировать, образуя бензилтозилат, или подвергаться сольволизу с участием растворителя:

3 Стереохимические аспекты моно-(SN1) и бимолекулярного нуклеофильного замещения (SN2).
Нуклеофильное замещение у насыщенного атома углерода (SN2) является одним из наиболее обширных и важных классов органических реакций. В этом процессе уходящая группа Х органического субстрата R-X, содержащего σ- связь С sp3 -X, замещается нуклеофильным реагентом Y: таким образом, что неподеленная пара электронов реагента становится электронной парой σ- связи C-Y продукта реакции, а электронная пара σ- связи C-X становится неподеленной парой электронов уходящей группы X, которую обычно называют нуклеофугом (fugitive-мимолетный, летучий, непрочный). К хорошим нуклеофугам относятся группы, уходящие в виде анионов сильных кислот - трифторметансульфонатная CF3SO3¯, фторсульфонатная FSO3¯, пара-толуолсульфонатная п -CH3C6H4SO3¯ (тозилатная), а также бромид- и иодид-анионы. Плохими нуклеофугами являются анионы слабых кислот, например, ацетат- и фторид - ионы.
Механизм процессов SN2 описывает синхронную одностадийную реакцию, переходное состояние которой представляет собой тригональную бипирамиду с пентакоординированным атомом углерода:

Главной движущей силой реакции является взаимодействие несвязывающей орбитали (неподеленной пары электронов) нуклеофила (ВЗМО) с нижней свободной (разрыхляющей) σ*- орбиталью связи C-X (НСМО). Согласованный механизм SN2 обуславливает общий второй кинетический порядок реакции, первый по субстрату и первый по нуклеофилу. Важной характеристикой SN2 –механизма является стереохимия реакции. Синхронный процесс SN2 должен быть стереоспецифическим, посколь-
ку, как показывают квантовохимические расчеты, атака нуклеофила происходит с тыла σ- связи C-X, т.е. со стороны, противоположной уходящей группе X.
Стереохимическим следствием этого должно быть т.н. вальденовское обращение конфигурации при хиральном (асимметрическом) атоме углерода, с образованием (при X=Y) энантиомера исходного субстрата, т.е. зеркального отражения его структуры. Хиральным (от греческого “хирос”- рука) может быть атом углерода в состоянии тетраэдрической (sp3) гибридизации, у которого все четыре заместителя разные, например, бромфторхлорметан:
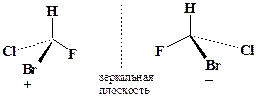
Уникальным свойством хиральных молекул является их оптическая активность, т.е. способность вращать плоскость поляризации плоскополяризованного света (оптическое вращение). Энантиомеры различаются по знаку вращения: (+) - правовращающий и (-) - левовращающий изомеры, а также по величине угла вращения. Если энантиомеры содержатся в смеси в эквимолекулярном соотношении, они образуют оптически неактивную смесь, называемую рацемической, или рацематом (±).
Абсолютная конфигурация энантиомера определяет истинное расположение атомов или групп у хирального центра и обозначается как R и S-конфигурации (от латинских слов ”rectus”- правый и “sinister”-левый). Для нахождения абсолютной конфигурации необходимо определение старшинства заместителя. Чем больше атомный номер, тем старше заместитель. Ряд уменьшения старшинства заместителей имеет вид: 1→2→3→4 (на вышеприведенном примере Br→Cl→F→H). Если у двух или трех заместителей первый атом одинаковый (например, углерод), то старшинство определяется по атомным номерам (массам) следующих за ним атомов и групп и характеру их связей. Например,

Для определения абсолютной конфигурации молекулу располагают так, чтобы наименьший по старшинству заместитель был направлен от наблюдателя:

Если в полученном изображении три старших заместителя при хиральном центре располагаются в порядке снижения старшинства по ходу часовой стрелки, это R-энантиомер, а если против - то мы имеем S-изомер. Знак оптического вращения прямо не связан с типом конформации и определяется экспериментально.
Как отмечалось выше, в переходном состоянии согласованной SN2 реакции степень координации центрального атома углерода возрастает до 5, а само переходное состояние представляет собой тригональную бипирамиду. Поэтому скорость реакции должна сильно зависеть от пространственных факторов, в первую очередь от объема заместителя у реакционного центра. Для алкилгалогенидов и алкилсульфонатов скорость уменьшается в ряду CH3 > C2H5 > RCH2 > R2CH > R3C. Таким образом, скорость SN2- замещения резко уменьшается при переходе от первичных к вторичным алкильным радикалам. Для третичных алкилгалогенидов (R3C-X) из-за стерических препятствий замещение возможно только по механизму SN1, сопровождаемому Е1- элиминированием.
Особо сильное пространственные препятствия создает трет.-бутильная группа (например, в неопентилбромиде (CH3)3C-CH2Br), как бы накрывающая зонтиком маршрут атаки нуклеофила,

Рис. Стерические препятствия SN2 замещению со стороны
метильных групп
а также трехмерный углеводородный каркас в производных 1-Х-бицикло- [2.2.1]гептана (А), бицикло[2.2.2]октана (В) и адамантана (С), который полностью блокирует подход нуклеофила с тыла рвущейся связи С-Х.

Как показывают квантовохимические расчеты, наиболее стабильной является плоская тригональная (sp2) структура третичного карбониевого иона с вакантной р -электронной орбиталью (в отсутствие ограничений, налагаемых пространственным каркасом)

Поэтому атака нуклеофилом такого карбокатиона равновероятна с обеих сторон от плоскости катиона и стереохимическим результатом ее будет образование рацемата, т.е. эквимолярной смеси R- и S-энантиомеров. Для вышеприведенных каркасных соединений плоская структура карбокатиона недостижима и атака нуклеофила происходит со стороны разорванной связи, давая продукт с сохранением конфигурации.
Это приводит к резкому замедлению реакции SN1. Так, относительные скорости сольволиза каркасных би- и трициклобромидов по сравнению с трет.-бутилбромидом, принятым за стандарт, (k отн=1), составляют: 10-13 (А), 10-6 (В) и 10-3 (С). Это показывает, что наиболее пространственно напряженным является каркас бицикло[2.2.1]гептана (норборнана) (А) и наименее – адамантана (С).
Для вторичных алкилгалогенидов и алкилсульфонатов в зависимости от природы субстрата, нуклеофила, уходящей группы и даже растворителя может реализоваться как SN2, так и SN1 -механизм нуклеофильного замещения, на что указывают стереохими-
ческие особенности этих реакций.
Например, при щелочном гидролизе α -фенилэтилхлорида в водном спирте образуется 17% α -фенилэтилового спирта с инверсией конфигурации и 83% рацемата:

Однако при действии на этот субстрат ацетат-иона в ацетоне количество инвертированного α -фенилэтилацетата составляет уже 65%.
При электрофильном катализе этой реакции солями серебра (способствующими образованию карбокатиона) образуется 87% продукта с сохранением конфигурации и 13% рацемата

Увеличение полярности растворителя, как правило, приводит к ускорению реакции SN1, т.к. в переходном состоянии происходит разделение зарядов, что стабилизирует переходное состояние за счет электростатической диполь-дипольной сольватации.
Чтобы объяснить наблюдаемые явления, С. Уинстейн предложил т.н. ионно-парный механизм мономолекулярного нуклеофильного замещения. Уинстейн считает, что ионизация молекулы с ковалентной связью R-X протекает через несколько последовательных стадий:

Первоначально при ионизации субстрата R-X образуется контактная ионная пара R+X¯, где катион и анион связаны силами электростатического притяжения и между ними нет разделяющих их молекул растворителя. Оба эти состояния субстрата R-X и R+X¯ подвергаются бимолекулярному нуклеофильному замещению с обращением конфигурации, поскольку атака тесной ионной пары нуклеофилом возможна только со стороны, противоположной уходящей группе Х¯.
При дальнейшем разделении компонентов ионной пары контактная пара превращается в сольватно-разделенную (“рыхлую”) ионную пару R+||X¯, в которой ионы разделены одной или несколькими молекулами растворителя и поэтому значительно слабее связаны друг с другом и более реакционноспособны, чем контактные.
При атаке нуклеофилом такой ионной пары возможно замещение нуклеофилом Y¯ уходящей группы Х¯ в составе пары, что обуславливает частичное сохранение конфигурации (при обратном переходе через тесную ионную пару к ковалентному продукту R-Y) и частичная рацемизация при атаке ионной пары R+||X¯ со стороны, противоположной уходящей группе Х. Наконец, при взаимодействии сольватированного катиона R+ с нуклеофилом Y¯ реализуется классический механизм SN1 с полной рацемизацией (или полным сохранением конфигурации для каркасных молекул).