Тип наследования
Известно, что данное заболеванием является генетическим, то есть передается по наследству. Изучение родословной позволит проследить хорею на протяжении 2-3 поколений, при этом 1 из родителей будет иметь эту патологию.
Для заболевания характерен аутосомно-доминантный тип наследования с полной пенетрантностью, то есть половина детей, которые появятся на свет от больного человека, будет страдать этим недугом.
Достаточно всего одного мутантного гена для проявления признаков патологии.
Формы заболевания
Неврологи выделяют основные формы:
· Классическая гиперкинетическая. Первые симптомы отмечают в 30-60 лет;
· Ювенильная. Симптоматика появляется до 20 лет;
· Психопатологическая.
Симптомы
Невозможно точно определить возраст, в котором у больного появятся первые симптомы заболевания. Это связывают с тем, что небольшие изменения в поведении могут возникнуть за многие годы до возникновения явной симптоматики. В результате диагноз обычно ставят к моменту появления хореической (непроизвольной) двигательной активности, нарушения координации движений.
Симптомы прогрессирующего заболевания:

· Снижение памяти;
· Человек не способен организовать свою работу;
· Затрудненная речь;
· Возникновение дистонических поз;
· Нарастание глазодвигательных движений;
· Серьезные нарушения координации движений.
Поздние стадии характеризуются появлением неразборчивой речи, затруднений при глотании, больной не в состоянии самостоятельно передвигаться, отмечают тотальную деменцию с распадом личности, на фоне которой возникают галлюцинации и психозы.
Течение болезни
За появление и течение хореи Гентингтона отвечает мутантный ген, расположенный на коротком плече 4 хромосомы. Продуктом данного гена становится крупный белок, получивший название гентингтин. Именно он ответственен за возникновения болезни, напрямую определяет сроки возникновения первых симптомов и тяжесть течения хореи. Из поколения в поколение характерно постепенное усугубление проявлений заболевания.
Гентингтин способен взаимодействовать с большим количеством прочих белковых соединений, выполняя ряд полезных биологических функций (предотвращает гибель клеток, участвует в передаче импульсов). Однако важной его особенностью является токсичность для нейронов.
Болезнь Гентингтона приводит к постепенной гибели нейронов, которые располагаются в области базальных ганглиев головного мозга. В результате также происходит снижение общего веса мозга вследствие разрушения белого вещества. Разрушение базальных ганглиев приводит к нарушениям движений и поведения больного.
Диагностика
Невролог способен поставить точный диагноз на основании типичной симптоматики, семейного анамнеза и результатов генетических анализов.
Во время заболевания происходит атрофия головки хвостатого ядра, в результате во время проведения МРТ или КТ на поздних стадиях можно отметить значительное расширение желудочков мозга. Однако использование методов функциональной нейровизуализации позволит определить изменения активности головного мозга до возникновения симптоматики.
Использование генетических методов позволяет точно определить хорею Гентингтона. Для этого производят забор крови. Положительный результат не будет подтверждением болезни, потому что первые симптомы могут возникнуть спустя много лет после анализа. Однако отрицательный результата будет свидетельствовать, что человек не заболеет в течение всей жизни.
Лечение

Хорея Гентингтона является неизлечимым заболеванием, однако существует терапия, способная облегчить некоторые симптомы. Непроизвольные движения и тревожные состояния стараются подавлять с помощью нейролептиков, к примеру, хлорпромазин или галоперидол. Возможно увеличение дозы до максимальных до тех пор, пока не появятся побочные эффекты (сонливость, паркинсонизм, депрессия).
Нейролептики последнего поколения (рисперидон, оланзапин) становятся препаратами выбора при хорее Гентингтона благодаря отсутствию тяжелых побочных эффектов и способности снижать тревожность, раздражительность и проявления паранойи. Однако они неэффективны на поздних стадиях болезни.
Анксиолитики, антидепрессанты также нашли широкое применение для терапии психоза, депрессии и раздражительности. Важно чтобы они были назначены исключительно на период, когда у пациента имеются в действительности данные симптомы.
С целью облегчить ригидность и гипокинезию мышечной ткани применяют противопаркинсонические средства. Широко назначают вальпроевую кислоту. Если у больного отмечаются нарушения поведения и психозы, то применяют атипичные антипсихотики.
 Обратите внимание также на материал о болезни Альцгеймера и старческом слабоумии.
Обратите внимание также на материал о болезни Альцгеймера и старческом слабоумии.
О стадиях болезни Паркинсона читайте на https://www.neuroplus.ru/bolezni/bolezn-parkinsona/diagnostika-i-lechenie-bolezni-parkinsona.html.
Прогноз
Прогрессирование хореи Гентингтона происходит на протяжении 15-20 лет. К терминальной стадии больной становится абсолютно беспомощным. Смерть наступает от осложнений заболевания, к примеру, присоединения инфекции. Достаточно часто причиной смерти является самоубийство больного.
Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона или Хантингтона) — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Эпидемиология[править | править вики-текст]
В настоящее время от хореи Хантингтона в США страдает около 7000 человек. Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000, и 1:1000000 среди остальных рас[1]. Название болезни дано в честь трёх поколений врачей, изучавших её в штате Коннектикут. В частности, считается, что заболевание названо в честь американского врача Джорджа Хантингтона, первым давшего его классическое описание[2][3].
Генетика[править | править вики-текст]
Ген HTT, присутствующий у всех людей, кодирует белок хантингтин (Htt). Ген HTT расположен на коротком плече 4-й хромосомы (4p16.3)[4]. Этот ген содержит в себе участок с повторяющейся последовательностью трёх азотистых оснований — цитозин-аденин-гуанин (т.е., ЦАГЦАГЦАГ...). Триплет ЦАГ кодирует аминокислоту глутамин, поэтому синтезируемый белок хантингтин содержит последовательность глутаминовых аминокислот, называемую полиглутаминовый тракт[5].
Количество ЦАГ триплетов различно у отдельных лиц и может изменяться с последующими поколениями. Если их становится больше 36, то синтезируется удлинённый полиглутаминовый тракт и происходит образование мутантного белка хантингтина (mHtt)[6], который оказывает токсичное действие на клетки и вызывает болезнь Хантингтона. Как правило, от числа ЦАГ повторов зависит степень повреждений, наличие около 60 % повторов сверх нормы вызывает появление симптомов в различном возрасте[4]. 36—40 повторов приводят к редуцированной пенетрантности формы этого заболевания, которая намного позже проявляется и медленнее прогрессирует. В некоторых случаях начало болезни может быть настолько поздним, что симптомы никогда не обнаруживаются[7]. При очень большом количестве повторов болезнь Хантингтона имеет полную пенетрантность и может проявиться до 20 лет, тогда болезнь классифицируется как ювенильная, акинетически-ригидная или Вестфаль варианты. Составляет приблизительно 7 % случаев болезни Хантингтона[8].
Мутантный ген был предположительно завезён в США в 1630 году двумя братьями, эмигрировавшими из Эссекса в Бостон[9][10].
Патогенез[править | править вики-текст]
Htt-белок взаимодействует с сотней других белков и, вероятно, выполняет множество биологических функций[11]. Механизм действия mHtt до конца не ясен, но известно, что он токсичен для некоторых типов клеток, особенно в головном мозге. В основном происходит поражение полосатого тела (стриатума), но при прогрессировании заболевания и другие области головного мозга значительно повреждаются[6]. Планирование и коррекция движений — основная функция полосатого тела, и нарушения в этой области провоцируют симптомы[6].
Функция Htt [править | править вики-текст]
Htt образуется во всех клетках млекопитающих. Наибольшая его концентрация — в головном мозге и яичках, а также в умеренных количествах в печени, сердце и лёгких[6]. Функция Htt у человека не ясна. Он взаимодействует с белками, участвующими в транскрипции, передаче сигнала в клетке и внутриклеточном транспорте[6][12]. Некоторые функции Htt обнаружены в экспериментальных моделях животных: играет важную роль в развитии эмбриона и связан с гибелью эмбриона при отсутствии белка[13]. Он также выступает в качестве анти-апоптозного агента, предотвращая запрограммированную гибель клеток, и контролирует образование нейротрофического фактора мозга (белок, защищающий нейроны и регулирующий их образование во время нейрогенеза). Если экспрессия Htt возрастает, выживаемость нервных клеток увеличивается и эффект mHtt уменьшается, наоборот, понижение экспрессии Htt даёт картину более типичную присутствию mHtt[13]. У людей разрушение нормального гена не приводит к болезни. В настоящее время считается, что болезнь вызывает не недостаточное образование Htt, а усиление токсического эффекта mHtt[6].
Клеточные изменения под действием mHtt [править | править вики-текст]
Под действием образовавшегося mHtt происходит множество изменений в клетке, что вызывает болезнь Хантингтона. Удлинение полиглутаминовой последовательности изменяет конформацию белка хантингтина и прочно соединяет его с другими белками[14]. Это приводит к агрегации хантингтина, при этом образуются так называемые внутриклеточные тельца включения[15]. Эти включения механически препятствуют движению везикул, содержащих нейромедиаторы, через цитоскелет, что нарушает передачу сигналов в нейронах[15]. Тельца включения обнаруживаются как в ядрах клеток, так и в цитоплазме. Некоторые эксперименты показали, что они могут быть токсичны для клеток, а другие — что тельца, наоборот, защищают нейрон от смерти, аккумулируя мутантный хантингтин, и именно неагрегированный белок токсичен[16].
Существует несколько путей, при которых mHtt вызывает гибель клеток. К ним относят: влияние на белки-шапероны; взаимодействие с каспазами, которые участвуют в апоптозе; токсическое действие глутамина на нервные клетки; нарушение выработки энергии в клетках и влияние на экспрессию генов. Токсическое действие mHtt значительно усиливается при взаимодействии с белком RASD2 (Rhes), который образуется преимущественно в стриатуме. RASD2 вызывает сумоляцию (SUMOylation) mHtt к образованию белковых сгустков и дезагрегации — исследования в культуре клеток показали, что сгустки менее токсичны, чем дезагрегированная форма[17].
Макроскопические изменения под действием mHtt [править | править вики-текст]

Область головного мозга, поражающаяся при болезни Хантингтона, — стриатум (розовым цветом)
Болезнь Хантингтона поражает специфические области мозга. Наиболее заметные ранние изменения затрагивают область базальных ганглиев, называемую полосатым телом, которое состоит из хвостатого ядра и скорлупы[6]. Другие повреждаемые области включают чёрную субстанцию, 3, 5 и 6 слои коры головного мозга, гиппокамп, клетки Пуркинье в мозжечке, боковые туберальные ядра гипоталамуса и часть таламуса[4]. Эти области получают повреждения в соответствии с их структурой и типами содержащихся в них нейронов, уменьшаясь в размерах в связи с гибелью клеток[4]. Звёздчатые нейроны полосатого тела наиболее уязвимы, особенно проецирующиеся в направлении поверхности бледного шара, вставочные и звёздчатые нейроны, проецирующиеся к центру бледного шара, получают меньше повреждений[4][18]. Болезнь Хантингтона также вызывает аномальное увеличение астроцитов[19].
Базальные ганглии — часть головного мозга, наиболее заметно повреждающиеся при болезни Хантингтона — играют ключевую роль в контроле движений и поведения. Их функция полностью не ясна, но современные теории предполагают, что они являются частью когнитивной исполнительной системы. Базальные ганглии в норме ингибируют большое число контуров (circuit), генерирующих специфические движения. Для инициации специфических движений кора посылает сигналы базальным ганглиям для снятия ингибирования. Повреждение базальных ганглиев может привести к снятию ингибирования или его постоянным неконтролируемым изменениям, что служит причиной затруднения начала движения или к их непроизвольной инициации, или движение может быть прервано до или после достижения желаемого результата. Накапливающиеся повреждения в этой области приводят к беспорядочным движениям, характерным для болезни Хантингтона[20].
Симптомы[править | править вики-текст]
Симптомы болезни Хантингтона могут проявиться в любом возрасте, но чаще это происходит в 35–44 года[21][22]. На ранних стадиях происходят небольшие изменения личности, когнитивных способностей и физических навыков[21]. Обычно первыми обнаруживают физические симптомы, так как когнитивные и психические расстройства не столь выражены в ранних стадиях[21]. Почти у всех пациентов болезнь Хантингтона в конечном итоге проявляется схожими физическими симптомами, но начало заболевания, прогрессирование и степень когнитивных и психических нарушений различаются у отдельных лиц[23][24].
Для начала заболевания наиболее характерна хорея — беспорядочные, неконтролируемые движения. Хорея в начале может проявляться в беспокойстве, небольших непроизвольных или незавершённых движениях, нарушении координации и замедлении скачкообразных движений глаз[21].
В самом начале обычно возникают проблемы из-за физических симптомов, которые выражаются в резких, внезапных и не поддающихся контролю движениях. В других случаях, наоборот, больной двигается слишком замедленно. Возникают нарушения координации движений, речь становится невнятной. Постепенно все функции, требующие мышечного контроля, нарушаются: человек начинает гримасничать, испытывает проблемы с жеванием и глотанием. Из-за быстрого движения глаз происходят нарушения сна. Обычно больной проходит через все стадии физического расстройства, однако влияние болезни на когнитивные функции у всех очень индивидуально. Чаще всего происходит расстройство абстрактного мышления, человек перестаёт быть способным планировать свои действия, следовать правилам, оценивать адекватность своих действий. Постепенно появляются проблемы с памятью, может возникнуть депрессия и паника, эмоциональный дефицит, эгоцентризм, агрессия, навязчивые идеи, проблемы с узнаванием других людей, гиперсексуальность и усиление вредных привычек, таких как алкоголизм или игромания.
Диагностика[править | править вики-текст]
Клинические методы [править | править вики-текст]
Физикальное обследование, иногда в сочетании с психологическим обследованием, позволяет определить область распространения болезни[21]. Медицинская визуализация (компьютерная томография (КТ), магнитно-резонансная томография (МРТ)) показывает только видимую атрофию мозга на прогрессирующей стадии заболевания. Методы функциональной нейровизуализации (фМРТ и позитронно-эмиссионная томография (ПЭТ)) могут показать изменения в активности мозга до появления клинических симптомов[4].
Генетические методы [править | править вики-текст]
Для проведения генетической диагностики болезни Хантингтона необходим забор крови с последующим определением количества повторов ЦАГ в каждом НТТ -аллеле[25]. Положительный результат не подтверждает диагноз, поскольку может быть получен за несколько лет до появления первых симптомов. Однако отрицательный результат однозначно свидетельствует об отсутствии вероятности развития болезни Хантингтона[4].
Эмбрионы [править | править вики-текст]
Эмбрионы, полученные в результате экстракорпорального оплодотворения, могут быть подвержены генетической диагностике болезни Хантингтона с применением преимплантационной генетической диагностики. При этом методе забирается одна клетка из 4–8-клеточного эмбриона и затем проверяется на генетическую патологию. Полученная информация может впоследствии быть использована при выборе здорового эмбриона для имплантации. Кроме того, возможна пренатальная диагностика для эмбриона или плода в утробе матери[26].
Дифференциальная диагностика [править | править вики-текст]
Около 90% диагнозов болезни Хантингтона, основанных на обнаружении типичных симптомов и семейном анамнезе, подтверждаются генетическим тестированием. Большинство других расстройств с аналогичными симптомами называют ХГ-подобными расстройствами (англ. HD-like disorders, HDL)[27]. Причины большинства HDL-заболеваний неизвестны. Известно лишь, что некоторые из них возникают в результате мутаций генов PRNP (HDL1), junctophilin 3 (HDL2), рецессивно наследуемого HTT гена (HDL3 — обнаружен у одной семьи и мало изучен) и гена, кодирующего ТАТА-связывающий белок (HDL4/SCA17)[27]. К другим заболеваниям с аутосомно-доминантным наследованием, которые схожи с болезнью Хантингтона, относят дентаторубро-паллидолюисовую атрофию и нейроферритинопатию[27].
Лечение[править | править вики-текст]

Химическая структура тетрабеназина, разрешённого для лечения болезни Хантингтона
Болезнь Хантингтона неизлечима, но существует лечение, способное облегчить некоторые симптомы.[28]
Тетрабеназин был разработан специально для уменьшения тяжести симптомов болезни Хантингтона[29], был утвержден в 2008 году в США[30]. Нейролептики и бензодиазепины помогают уменьшить проявления хореи[22]. Амантадин и ремацемид находятся в стадии исследования, но показали положительные результаты[31]. Для облегчения гипокинезии и ригидности мышц назначают противопаркинсонические лекарства, для облегчения миоклонической гиперкинезии — вальпроевую кислоту[22].
Для устранения депрессии применяют селективные ингибиторы обратного захвата серотонина и миртазапин, а при психозах и нарушениях поведения назначают атипичные антипсихотики[32].
В настоящий момент ведутся активные исследования по разработке способа лечения, исследуются потенциальные направления для лечения болезни Хантингтона[33].




| Генетические нарушения | |
| Тип наследования | |
| Частота наследования | |
| Возраст манифестации | |
| Основные клинические проявления | |
| Специфическая лабораторная диагностика | |
| Специфические медицинские мероприятия для облегчения лечения | |
| Дополнительные сведения |
Синдром Туре́тта (болезнь Туретта, синдром Жиля де ла Туретта) — генетически обусловленное расстройство центральной нервной системы, которое проявляется в детском возрасте и характеризуется множественными моторными тиками и как минимум одним вокальным или механическим тиком.
Ранее синдром Туретта считался редким и странным синдромом, ассоциируемым с выкрикиванием нецензурных слов или социально неуместных и оскорбительных высказываний (копролалия). Однако этот симптом присутствует только у меньшего числа людей с синдромом Туретта[1]. У людей с синдромом Туретта уровень интеллекта и продолжительность жизни в норме. Степень выраженности тиков уменьшается у большинства детей, когда у них завершается подростковый возраст, а тяжёлая степень синдрома Туретта в зрелом возрасте встречается редко. Известные люди с синдромом Туретта встречаются во всех сферах жизни[2].
Генетические и экологические факторы играют определённую роль в этиологии синдрома Туретта, но точные причины заболевания неизвестны. В большинстве случаев лечение не требуется. Не существует эффективных лекарственных средств для каждого случая тиков, но использование лекарств и методов лечения, которые облегчают состояние больного, оправдано. Обучение, разъяснение этого заболевания и психологическая поддержка больных — важная часть плана лечения[3].
Эпоним был предложен Жаном Мартеном Шарко в честь своего ученика, Жиля де ла Туретта, французского врача и невролога, который опубликовал отчёт о 9 больных с синдромом Туретта в 1885 году.
Содержание
[скрыть]
· 1История
o 1.1Донаучный период (Средневековье)
o 1.2XIX век
· 2Эпидемиология
· 3Этиология
· 4Классификация
· 5Клиническая картина
· 6Лечение
· 7Примечания
· 8Ссылки
История[править | править вики-текст]
Донаучный период (Средневековье) [править | править вики-текст]
Впервые состояние, похожее на синдром Туретта, было описано в 1489 в «Молоте ведьм» (лат. «Malleus Maleficarum») Г. Крамером и Я. Шпренгером. В книге описан священник, имевший моторные и вокальные тики и считавшийся «одержимым»[4].
XIX век [править | править вики-текст]

Ж. М. Г. Итар — французский медик, один из основателей детской психиатрии, впервые с научной точки зрения описавший синдром Туретта[5]
В 1825 году Ж. М. Г. Итар, в то время — руководитель Королевского института глухонемоты (теперь — Парижский национальный институт молодых людей с глухотой (англ.)русск.) опубликовал статью фр. «Mémoire sur quelques fonctions involontaires des appareils de la locomotion, de la préhension et de la voix» («Научное исследование некоторых непроизвольных функций системы органов, связанной с движением, хватанием и голосом»), в которой описал 10 случаев синдрома Туретта (7 из больных — мужчины)[4]. Среди больных он описывает некую маркизу Дампьер, парижскую аристократку и довольно влиятельную особу своего времени[6]. Она страдала от малоописанного тогда состояния, идентифицированного современными учёными как копролалия. Дама совершала вокальные тики в виде непристойной брани, что вступало в резкий контраст с её происхождением, интеллектом и благородными манерами[4].
В 1861 году важный вклад в исследование синдрома Туретта сделал А. Труссо, описавший несколько случаев в своём учебнике по клинической медицине и терапии[4].
Эпидемиология[править | править вики-текст]
Синдром Туретта в настоящее время не считается редкой болезнью, но он не всегда может быть диагностирован верно, потому что большинство случаев протекает в лёгкой форме. От 1 до 10 детей из 1000 имеют синдром Туретта[7]; более, чем 10 на 1000 человек имеют тиковые расстройства[8][9]. По другим данным, распространённость синдрома Туретта среди населения в целом составляет 3—5 случая на 10 000 человек[10]. Преобладают лица мужского пола (3:1)[10].
Этиология[править | править вики-текст]
Этиология и точная причина синдрома Туретта неизвестны, но установлена связь с генетическими и экологическими факторами[11]. Генетические исследования показали, что подавляющее большинство случаев синдрома Туретта передаются по наследству, хотя точный механизм наследования пока не определён[12] и специфический ген не выявлен.[13] В некоторых случаях синдром Туретта спорадический, то есть не унаследован от родителей[14]. Другие расстройства в виде тиков, не связанные с синдромом Туретта, называют туреттизмами [15].
Человек с синдромом Туретта имеет около 50 % вероятность передачи гена (генов) одному из своих детей, но синдром Туретта — состояние с вариабельной экспрессией генов и с неполной пенетрантностью[16]. Таким образом, не у каждого, кто унаследовал данный генетический дефект, проявятся симптомы; даже у близких родственников могут проявляться симптомы различной степени тяжести или их вообще может не быть. Ген (гены) может экспрессироваться в синдром Туретта как тик лёгкой степени (преходящий или хронический тики) или как обсессивно-компульсивные симптомы без тиков. Лишь незначительная часть детей, которые унаследовали ген(ы), имеют симптомы, требующие медицинского внимания[17]. Пол, судя по всему, влияет на экспрессию дефектного гена: у мужчины чаще проявляются тики, чем у женщин[18].
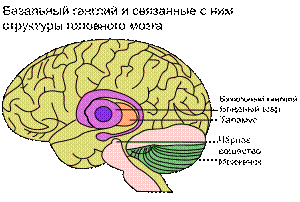
Структуры головного мозга, связанные с развитием синдрома Туретта
Негенетические, экологические, инфекционные или психосоциальные факторы, не вызывающие синдром Туретта, способны влиять на его тяжесть[13]. Аутоиммунные процессы могут провоцировать возникновение тиков и их обострение в некоторых случаях. В 1998 году группа американских учёных Национального института психического здоровья выдвинула гипотезу, что обсессивно-компульсивные расстройства и тики могут возникать у группы детей в результате постстрептококкового аутоиммунного процесса[19]. Дети, у которых обнаруживают 5 диагностических критериев, классифицируются в соответствии с этой гипотезой как имеющие детские аутоиммунные нервно-психические расстройства, ассоциированные со стрептококковой инфекцией (англ. аббревиатура PANDAS)[20]. Эта спорная гипотеза находится в центре внимания клинических и лабораторных исследований, но остаётся недоказанной[21][22].
Тики, как полагают, — результат дисфункции таламуса, базальных ганглиев и лобных долей[11]. Нейроанатомические модели объясняют причастность к данному синдрому сбоев в нейронных связях корковых и подкорковых структур головного мозга[13], а технологии нейровизуализации объясняют причастность базальных ганглиев и лобных извилин[23].
Некоторые формы обсессивно-компульсивных расстройств могут быть генетически связаны с синдромом Туретта[24][25].
Была также предложена теория, утверждающая, что недостаток магния в организме и вызванные этим метаболические нарушения могут быть одной из причин синдрома Туретта и некоторых связанных с ним коморбидных состояний[26], при этом прием соединений магния и витамина B6 может улучшать состояние многих больных[27].
Классификация[править | править вики-текст]
Тики возникают внезапно в виде повторяющихся, однообразных, неритмичных движений (моторные тики) и высказываний (звуковые тики) с участием отдельных групп мышц[28].
Синдром Туретта — один из видов тиковых нарушений, которые классифицируются согласно «DSM-IV» в зависимости от типа (моторные или звуковые тики) и продолжительности (преходящий или хронический). Преходящее тиковое расстройство состоит из множественных двигательных тиков, звукового тика или обоих видов тика с продолжительностью от 4 недель до 12 месяцев. Хронические тиковые расстройства могут быть одиночные или множественные, моторные или звуковые тики (но не оба сразу), которые присутствуют более года[28]. Синдром Туретта диагностируется, когда множественные моторные тики и, по крайней мере, один звуковой тик присутствуют более года[29].
Тиковые расстройства определяются аналогично Всемирной организации здравоохранения (по МКБ-10)[30]. В международной классификации болезней F95.2 — «комбинированное голосовое и множественное двигательное тикозное расстройство». Для постановки диагноза синдрома Туретта по МКБ-10 состояние должно соответствовать следующим критериям[31]:
· A) множественные двигательные тики и один и более голосовых, представленых некоторое время тому назад, не обязательно непрерывно;
· B) тики должны наблюдаться много раз в течение дня, почти каждый день, расстройство длится дольше 1 года, продолжительность ремиссий не достигает 2 месяцев;
· C) начало в возрасте до 18 лет.
Клиническая картина[править | править вики-текст]
Тики — движения и звуки, «которые возникают периодически и непредсказуемо на фоне нормальной двигательной активности»[32], похожие на «отклонение нормального поведения»[18]. Тики, связанные с синдромом Туретта, различаются по количеству, частоте, тяжести и анатомической локализации. Эмоциональные переживания увеличивают или уменьшают выраженность и частоту тиков у каждого больного индивидуально. Также тики у некоторых больных протекают «приступ за приступом»[28].
Копролалия (спонтанное высказывание социально нежелательных или запрещённых слов или фраз) — наиболее распространённый симптом болезни Туретта, но это не патогномонично для диагностики синдрома, так как только у около 10 % больных он проявляется[33]. Эхолалия (повторение чужих слов) и палилалия (повторение одного собственного слова) возникают реже[28], а наиболее часто в начале возникают моторные и вокальные тики, соответственно, в виде моргания глаз и кашля[34].

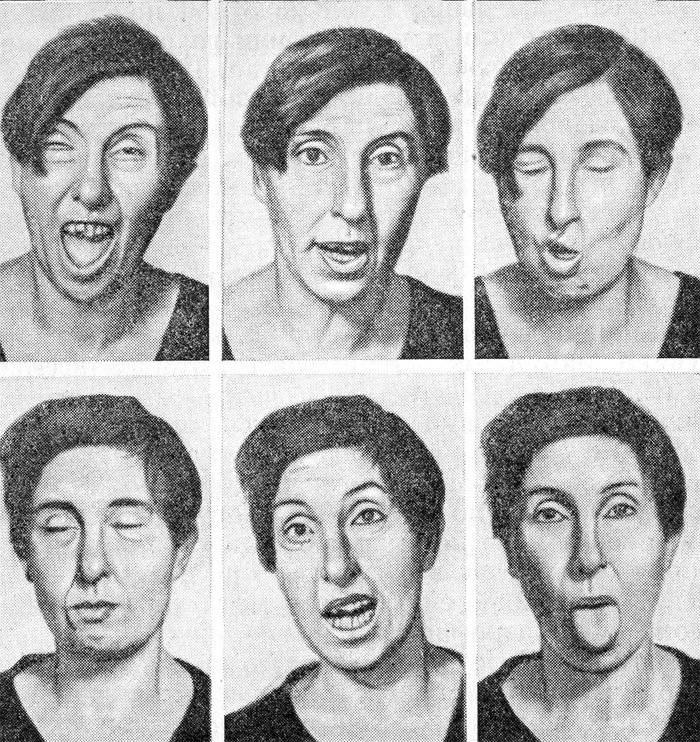
Примеры моторного тика
В отличие от патологических движений других двигательных расстройств (например, хорея, дистонии, миоклонус и дискинезии), тики при синдроме Туретта однообразные, временно подавляемые, неритмичные и часто им предшествует непреодолимое побуждение[35]. Непосредственно перед началом тика большинство людей с синдромом Туретта испытывают сильное побуждение[36][37], как при необходимости чихнуть или почесать зудящую кожу. Больные описывают позыв к тикам как нарастание напряжённости, давления или энергии[37][38], которую они сознательно освобождают, так как им «необходимо»[39] облегчить ощущения[37] или «вернуть себе хорошее самочувствие»[24][39]. Примеры такого состояния: ощущение инородного тела в горле или ограниченный дискомфорт в плечах, что приводит к необходимости откашливаться или пожать плечами. Фактически тик может ощущаться как освобождение этой напряжённости или ощущения, как и царапанье зудящей кожи. Другой пример — мигание для облегчения неприятного ощущения в глазах. Эти побуждения и ощущения, предшествующие появлению движений или вокализму как тикам, называются «продромальные сенсорные феномены» или продромальные позывы. Так как позывы предшествуют, тики характеризуются как полудобровольные[32]; они могут восприниматься как «добровольный», подавляемый ответ на непреодолимый продромальный позыв[33]. Опубликованы описания тиков при синдроме Туретта, определяющие сенсорные феномены как основной симптом заболевания, даже если они не включены в диагностические критерии[38][40][41].
Лечение[править | править вики-текст]

Клонидин, одно из лекарств, используемых при синдроме Туретта.
Лечение синдрома Туретта направлено на оказание помощи пациентам в управлении наиболее проблемными симптомами[13]. В большинстве случаев синдром Туретта протекает в лёгкой форме и не требует фармакологического лечения. Лечение (если оно требуется) направлено на устранение тиков и сопутствующих состояний; последние при возникновении часто становятся более проблемными, чем тики. Не все люди с тиками имеют сопутствующие состояния, но если они возникают, то лечение фокусируется на них. Не существует никакого лечения синдрома Туретта и нет лекарств, которые действовали бы универсально для всех людей без значительных побочных эффектов. Понимание больными своего заболевания позволяет эффективнее управлять тиковыми расстройствами[13]. Управление симптомами синдрома Туретта включает фармакологическую и психологическую терапию, правильное поведение. Фармакологическое лечение предназначено для тяжёлых симптомов, другие методы лечения (например, поддерживающая психотерапия и когнитивно-поведенческая терапия), могут помочь избежать или смягчить депрессию и социальную изоляцию. Обучение пациента, семьи и окружающих людей (например, друзей, школы) — одна из ключевых стратегий лечения и, может быть, это всё, что требуется в лёгких случаях[13][42].
Строение галоперидола. Галоперидол — антипсихотическое средство, используемое иногда при лечении тяжёлых случаев синдрома Туретта.
Лекарства применяют, когда симптомы мешают нормальной жизнедеятельности больного[17]. Классы лекарств с наиболее доказанной эффективностью в лечении тиков — типичные и атипичные антипсихотические препараты, включающие Рисперидон, Зипрасидон, Галоперидол (Галдол), Пимозид и Флуфеназин — могут вызывать долгие и кратковременные побочные эффекты. Антигипертензивные средства клонидин и гуанфацин также используются для лечения тиков; исследования показали переменную эффективность, но эффект ниже, чем у антипсихотических средств[1]. Исследования по применении метоклопрамида (церукала)[43] при синдроме Туретта (генерализованные тики и вокализация у детей)показали положительные результаты, однако медики отмечают, что для применения в педиатрической практике необходимы более масштабные испытания[44].
При навязчивостях, проблемах с концентрацией и депрессии, сопутствующей синдром Туретта, применяются трициклические антидепрессанты, СИОЗС (флуоксетин), а также литий[10].
Синдром Туретта является неврологическим заболеванием с генетической предрасположенностью, которое проявляется в раннем детстве или в подростковом возрасте до 18 лет. Синдром Туретта определяется наличием нескольких двигательных тиков и одним механическим или вокальным тиком, продолжительность которых должна быть больше года. Первые симптомы обычно являются непроизвольными движениями (тик) лица, руки, ноги или туловища. Эти тики частые, повторяющиеся и быстрые. Наиболее распространенным является симптомом лицевого тика (моргание глазом, дрожание носа, гримаса), и возможны другие добавочные тики шеи, туловища и конечностей.
Первый случай заболевания синдромом Туретта был зафиксирован в 1825 году доктором Итардом. Он описал патологию, схожую с синдромом, у довольно известного маркиза Де Дампьера - французского придворного, который запомнился миру наличием множественных двигательных и вокальных тиков. В 1885 году французский невролог Жорж Жиль Де ля Туретт, по заданию своего учителя известного французского невролога Шарко наблюдал в своей больнице девять пациентов с подобными симптомами, которые он систематизировал в одно заболевание описав его в своих трудах. В дальнейшем синдром стал носить его имя. Раньше людей с этим заболеванием называли бесноватыми. Из самых известных знаменитых людей - футболист Дэвид Бекхэм страдает легкой степенью синдрома Туретта в виде копролалии (выкрикивание бранных слов без повода).
Среди пациентов с синдромом гораздо чаще встречается психические расстройства: шизофре