Основой качественного спектрального анализа является то, что линейчатый спектр излучения каждого химического элемента является характеристичным. Задача качественного спектрального анализа сводится к отысканию линий определяемого элемента в спектре пробы. Принадлежность линии данному элементу устанавливается по длине волны и интенсивности линии. Расшифровывают спектры и определяют длину волн спектральных линий с помощью спектров сравнения, чаще всего спектра железа, имеющего характерные группы спектральных линий в разных областях длин волн. Спектр анализируемого вещества обычно фотографируют над спектром железа. Для определения длины волны какой-то линии в анализируемом спектре измеряют расстояние от этой линии до ближайших к ней справа и слева линий спектра железа, длины волн которых точно известны, и рассчитывают длину волны интересующей линии по интерполяционным формулам.
 По атласу определяют длины волн линий λ1 и λ2 в спектре железа, уточняя их значения по таблицам, а затем измеряют расстояние «а» между ними в мм на экране спектропроектора и расстояние от исследуемой линии до одной из выбранных опорных линий железа, т.е. b1 и b2.Тогда a/b2=λ1-λ2/λ1-λх=∆ λ2,1/∆ λ2,х Искомая длина волны равна λх = λ2 - b2/a*(λ2-λ1)
По атласу определяют длины волн линий λ1 и λ2 в спектре железа, уточняя их значения по таблицам, а затем измеряют расстояние «а» между ними в мм на экране спектропроектора и расстояние от исследуемой линии до одной из выбранных опорных линий железа, т.е. b1 и b2.Тогда a/b2=λ1-λ2/λ1-λх=∆ λ2,1/∆ λ2,х Искомая длина волны равна λх = λ2 - b2/a*(λ2-λ1)
Таблицы спектральных линий содержат перечень линий спектров элементов с указанием длин волн и интенсивностей спектральных линий в относительных единицах. Атласы спектральных линий содержат увеличенные фотографические или графические изображения спектров элементов, на которых указаны длины волн линий.Однако определения значения длины волны еще недостаточно, чтобы однозначно идентифицировать линию. Это связано с тем, что если отсутствие аналитической линии определяемого элемента в спектре пробы гарантирует отсутствие других линий этого элемента в спектре и его самого в пробе, то наличие линии с длиной волны, характерной для последней линии какого-либо элемента, еще не означает, что линия действительно принадлежит этому элементу и, следовательно, элемент присутствует в пробе. Основной причиной этого является так называемое наложение спектральных линий, связанное с недостаточной разрешающей способностью используемых спектральных приборов. Исключение возможных наложений спектральных линий проводят с использованием атласов спектров и таблиц спектральных линий.
Спектральным анализом качественно можно определить около 80 элементов. В связи с большой чувствительностью спектрального анализа существует опасность «переоткрывания» тех или иных элементов, попавших в пробу в результате случайных загрязнений.
Количественный спектральный анализ основан на зависимости интенсивности спектральных линий определяемых элементов от концентрации этих элементов в анализируемой пробе. В атомно-эмиссионном анализе зависимость интенсивности I линий характеристического спектра от концентрации С элемента в пробе выражается эмпирической формулой Ломакина–Шейбе: I=aCb, где а – коэф., зависящий от режима работы источника возбуждения, его стабильности, температуры и т.п.; b – коэффициент, учитывающий реабсор6цию или самопоглощение, т.е. поглощение квантов, испускаемых возбужденными атомами, атомами невозбужденного вещества; I- интенсивность спектральной линии определяемого вещества. Ф-лу используют, когда а и b определяют опытным путем для каждого случая!!!!
В зависимости от точности оценки интенсивности спектральных линий: -визуальные (оценка интенсивности проводиться на глаз); -фотографические (по величине почернения фотопластинки в месте расположения на ней спектральной линии); -фотоэлектрические (интенсивность измеряется с помощью фотоэлемента или ФЭУ).
Методы количественного атомно-эмиссионного анализа подразделяются на: полуколичественные (метод сравнения, гомологических пар, метод появления и исчезновения спектральных линий) и точные количественные (фотографический метод, трех стандартов, постоянного графика, добавок).
Полуколичественный анализ c использованием фотографической регистрации спектров методом сравнения состоит в том, что на одной и той же фотопластинке в одних и тек же условиях фотографируют спектры ряда стандартов с известным содержанием определяемых элементов и спектры анализируемых проб. Визуальным сравнением почернения спектральных линий в стандартных растворах и пробах определяют пределы содержания элементов в пробах. Точность анализа будет определяться тем, насколько сильно различается содержание определяемого элемента в стандартах – чем меньше разность концентраций между стандартами, тем точнее можно определить содержание анализируемого элемента в пробах.
При анализе металлов и сплавов, метод гомологических пар: В спектре элемента, определяемого в анализируемой пробе, выбирают ряд линий, называемых аналитическими. В спектре основного компонента анализируемой пробы (в сталях – Fe) или в спектреимеющегося в ней элемента или специально введенной добавки выбирают линии сравнения – так называемые гомологические линии. Гомологические линии должны удовлетворять следующим условиям:1)иметь как можно более близкие энергии возбуждения;2)принадлежать спектрам элементов, имеющих близкие к определяемому элементу энергии ионизации, температуры плавления и кипения, а также близкие химические свойства;3)отличаться от сравниваемых линий определяемого элемента по длинам волн не более чем на 100 А°, а по интенсивности не более, чем в 10 раз.
Исходя из предварительных исследований или используя литературные данные устанавливают, при каких содержаниях определяемого элемента в анализируемой пробе интенсивности аналитических линий и линий сравнения – т.н. гомологических пар – будут равны, и используют далее эти данные при проведении анализа.
Полуколичественный метод появления и усиления спектральных линий в атомно-эмиссионном анализе с помощью эталонов устанавливают, при каких концентрациях появляются или исчезают те или иные чувствительные линии элементов, а затем, опираясь на полученные данные, находят ориентировочное содержание элементов в пробе. При этом используется несколько аналитических линий определяемого элемента разной интенсивности.
Наиболее распространенным методом количественного анализа является метод трех стандартов: на одной пластинке фотографируют спектры анализируемых проб и спектры не менее трех стандартов. На микрофотометре измеряют почернение аналитических пар линий и строят градировочный график S — 1gС для стандартов, который является графиком данной фотопластинки и используется для определения неизвестной концентрации данного компонента в анализируемых пробах. Этот метод универсален и прост. Его удобно применять при одновременном анализе большого количества проб. Но при подборе условий анализа необходимо добиваться, чтобы почернения спектральных линий аналитических пар для всей определяемой области концентраций лежали в области нормальных почернений. Метод трех стандартом — самый точный метод, но имеет недостатки: длительность и необходимость получать для каждой пластинки свой калибровочный график, а также то, что погрешность определения может возрастать вследствие изменения свойств стандартов с течением времени. Модификацией метода трех стандартов является метод одного стандарта: для построения градуировочного графика используют один и тот же раствор стандарта, который распыляют в течение разного времени и измеряют суммарную интенсивность в течение этого времени распыления. При этом подбирают такие условия, при которых интенсивность аналитического сигнала пропорциональна общему количеству определяемого элемента, вводимого в источник атомизации и возбуждения. Чаще всего метод одного эталона применяется при анализе сплавов. Применяют также метод постоянного графика, сущность которого состоит в использовании заранее построенного по большому числу стандартов градуировочного графика в координатах lg(Iпр/Iосн.) — lg С. Для учета свойств фотопластинки, используемой при проведении каждого анализа, при расчете интенсивностей Inp/Iосн применяют коэффициент контрастности фотопластинки, который определяют по характеристической кривой фотоэмульсии.
При анализе проб сложного состава, исследовании чистых и сверхчистых веществ или при отсутствии материала соответствующей чистоты и невозможности по этой причине приготовить стандарты широко применяется метод добавок. При анализе по методу добавок пробу обычно переводят в раствор и делят его на несколько частей. Затем в каждую часть добавляют различные, но известные количества определяемого элемента, снимают спектры и определяют интенсивность спектральных линий. При расчете результатов анализа строят график, основываясь на том, что в области малых концентраций достаточно хорошо выполняется линейная зависимость Iпр/Iосн=аС, т.к. с уменьшением концентрации коэффициент реабсорбции b=1. В этом случае, если Сх — неизвестная концентрация, а Сст. —концентрация введенного стандарта (добавки), то Iпp/Iосн=а(Сх+Сст.). Сст. имеет ряд значений. Обычно Сст.2 = 2Сст.1; Сст.3 = 3Сст.1 и т.д. Откладывая на графике Іпр./Іосн. как функцию Сст. получают прямую, пересекающую ось ординат при Сст.=0. Экстраполяция этой прямой до пересечения с осью абсцисс дает на оси Сст. отрезок, равный Сх. Метод добавок очень ценен, т.к. применим для образцов, для которых неизвестен состав основы. Однако при его применении должны выполняться следующие условия: -градировочный график должен быть линейным; -добавляемый элемент должен находиться в той же химической форме, что и определяемый элемент; -добавка должна быть равномерно распределена в пробе.

Сх Сст.1 Сст.2 Сст.3
Метод добавок является надежным контрольным методом для установления правильности анализа.
Атомно-абсорбционная спектроскопия – физические основы метода, особенности атомизации вещества и возбуждения спектров поглощения. Анализаторы и детекторы излучения, применяемые в атомно-абсорбционной спектроскопии.
Атомно-абсорбционная спектроскопия основана на способности анализируемого вещества поглощать характеристическое для каждого вида атомов электромагнитное излучение, энергия которого приводит к переходу внешних электронов этих атомов из основного энергетического состояния на возбужденные энергетические уровни. При этом интенсивность возбуждающего излучения понижается в соответствии с основным законом светопоглощения – законом Бугера-Ламберта-Бера, из которого следует, что зависимость между светопоглощением и концентрацией поглощающих атомов линейна, а температура атомизатора на поглощение света не влияет.
 ,где
,где  - интенсивность на данной длине волны,
- интенсивность на данной длине волны, 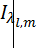 - интенсивность излучения прошедшего через слой,
- интенсивность излучения прошедшего через слой,
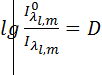 -оптическая плотность
-оптическая плотность
Коэффициент поглощения k пропорционален вероятности перехода внешних электронов на возбужденные уровни. Наиболее высокие значения коэффициента поглощения обычно соответствуют переходу электрона с основного на наиболее близкий к нему уровень (резонансная линия). Атомизация пробы в атомно-абсорбционной спектроскопии проводится при температуре 2000-3000 оС. В этом температурном интервале более 90% атомов находятся в невозбужденном состоянии и остальные компоненты атомизированной пробы не могут его изменить, следовательно, не могут повлиять на величину атомного поглощения. Это, а также небольшое количество линий в спектре поглощения атомов в данных условиях обусловливает высокую избирательность атомно-абсорбционного метода. Для точного измерения величины атомного поглощения А необходимо соблюдение двух условий, сформулированных Уолшем:
1) lЕмакс. = lDмакс. - длина волны, соответствующая максимальной интенсивности излучения используемого источника, должна быть равна длине волны максимального поглощения атомных паров lАмакс.
2) dА ≥ 2dЕ- полуширина dА линии поглощения атомных паров должна быть по крайней мере в два раза больше полуширины dЕ линии испускания источника.
Если первое условие Уолша не выполняется, поглощение электромагнитного излучения атомами анализируемого вещества вообще не происходит. Если не выполняется второе условие Уолша, то атомами определяемого элемента поглощается лишь небольшая часть излучения источника, так как контур линии эмиссии шире контура линии поглощения. Это приводит к резкому ухудшению чувствительности анализа.
Атомизатор - это устройство, необходимое для перевода анализируемой пробы в атомные пары. В отличие от атомно-эмиссионного анализа, где для атомизации пробы могут быть использованы различные методы (нагревание, бомбардировка ускоренными частицами, воздействие мощным потоком электромагнитного излучения и т.п.), в атомно-абсорбционном анализе атомизация пробы достигается ее нагреванием до 2000-3000 оС. Атомизация вещества анализируемой пробы в атомно-абсорбционных спектрометрах проводится в атомизаторах двух типов - пламенных и непламенных (электротермических). Пламенные атомизаторы в настоящее время используются чаще, чем непламенные. Атомизация вещества в них происходит под действием пламени, т.е. низкотемпературной плазмы, в которой определенный температурный баланс поддерживается за счет протекающих реакций окисления горючих газов в окислительной среде. Используемые смеси горючих газов с окислителями должны обеспечивать выполнение следующих условий, часто противоречащих друг другу: 1. Пламя должно быть высокопрозрачным 2. Собственное излучение пламени должно быть слабым, так как модулятор устраняет влияние этого излучения лишь до определенной степени. 3. Эффективность атомизации должна быть как можно более высокой. Этому обычно способствует наличие в пламени углеводородных радикалов продуктов горения, повышающих температуру пламени. 4. Степень ионизации, которая быстро возрастает с повышением температуры, должна быть низкой. Чувствительность атомно-абсорбционного анализа с атомизацией в пламени ограничена происходящими в нем побочными реакциями и кратким временем пребывания частиц анализируемой пробы (~ 10-3 с). Влияние этих факторов на чувствительность анализа резко снижается при проведении непламенной (электротермической) атомизации, которая осуществляется в специальной печи (т.е. в ограниченном объеме) и в инертной атмосфере, что снижает вероятность протекания побочных реакций. Такая печь должна соответствовать следующим требованиям:- быстро (в течение нескольких секунд) и воспроизводимо нагреваться дон ужной температуры (от 50 до 3000 оС);- иметь небольшую массу (для снижения тепловойи нерции);- иметь рабочую полость с изотермическим температурным режимом, иначе проба конденсируется на более холодных участках стенок, что приводит к заражению последующих анализируемых проб;- иметь высокую коррозионную и термическую стойкость во всем рабочем интервале температур.