Получение спиртов из галогеноуглеводородов[править | править вики-текст]
Галогенпроизводные углеводородов под действием оснований трансформируются с образованием спиртов (реакция нуклеофильного замещения).
Обычно, первичные и вторичные галогенуглеводороды вступают в реакцию по одностадийному SN2 механизму[3]. Пример — гидролиз бромэтана:
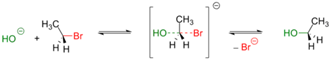
Реакции такого типа, обычно, происходят стереоспецифично — с обращением геометрической конфигурации исходного вещества[3]. Реакционная способность алкилгалогенидов уменьшается при переходе от производных йода к производным фтора [4] При этом фторпроизводные устойчивы к нуклеофильному замещению в обычных условиях и практически не используются для получения спиртов.
Первичные хлоралканы удовлетворительно гидролизуются под действием водного раствора щёлочи при нагревании[5]:

Для реакций, протекающих по SN2 механизму, используют только полярные растворители, причем скорость превращения возрастает при использовании вместо протонных растворителей (например: вода или спирт) апротонные (например: диметилсульфоксид); при этом в апротонных растворителях нуклеофильность уходящих групп будет иной[3]:

Третичные и в меньшей степени вторичные галогенуглеводороды гидролизуются по двухстадийному SN1 механизму[3]:
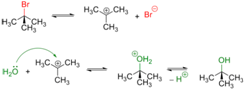
Реакция, протекающая по SN1 механизму проводят в полярных протонных растворителях, чаще всего воде или водном растворе метилового или этилового спирта.
Из-за устойчивости карбкатиона по такому механизму гидролизуются галогеналкены:

Так как в процессе реакции образуется карбкатион, его атака (в идеальных условиях без учета фактора влияния заместителей) нуклеофилом может происходить с обеих сторон, что приводит к рацемизации образующегося продукта.
Для высокореакционных реагентов используют мягкое замещение с использованием соединений одновалентного серебра или двухвалентной ртути[5]:
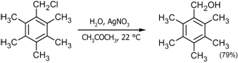

В современной лабораторной практике описанные выше реакции сольволиза проводят достаточно редко, так как спирты — более доступные полупродукты — являются исходным объектом для синтеза галогенпроизводных. Кроме того, следует помнить, что помимо изменения стереохимии исходных компонентов, реакции замещения конкурируют с элиминированием, а также перегруппировками, что часто приводит к нежелательным продуктам[3]:


В то же время существует достаточно новый метод превращения в спирты алкилгалогенидов действием на последние супероксида калия в средедиметилсульфоксида в присутствии 18-краун-6 полиэфира, при этом происходит практически полное геометрическое обращение[2]:

|
Получение спиртов из алкенов[править | править вики-текст]
Гидратация алкенов[править | править вики-текст]
Кислотная гидратация алкенов исторически была первым синтетическим методом получения спиртов (см. подраздел «История открытия спиртов»).
Общий механизм процесса (реакция электрофильного присоединения AdE2) представлен ниже[6]:


Присоединение происходит по правилу Марковникова.
В случае использования серной кислоты в качестве катализатора промежуточным продуктом является эфир серной кислоты (R-CH(OSO2OH)-CH3), который в условиях реакции полностью гидролизуется до спирта[6].
Для проведения реакции кроме серной кислоты используют и другие реагенты: смесь муравьиной и каталитического количества серной кислоты (в отдельных случаях позволяет добиться стереоспецифичности), смесь муравьиной и хлорной кислоты, трифторуксуную кислоту и др[7].
Реакции вторичных алкенов, вследствие перегруппировок карбокатионов, часто приводят к образованию смеси продуктов, что затрудняет их использование для получения вторичных спиртов[8]:

В лабораторной практике метод кислотной гидратации применим весьма ограниченно как из-за перспективы получения смеси продуктов, так и низких выходов. Чаще его используют для получения третичных спиртов, но и в этом случае выход, обычно, не превышает 40-45 %[8]:


|
В промышленности, помимо жидкофазной используют прямую газофазную гидратацию алкенов. В качестве катализаторов используется фосфорная кислота на твердом носителе при 200—300 °C и давлении 2-8 МПа; при этом выход спиртов достигает 95 %[9]:


28. Фенолы. Кислотность фенола. SЕ-реакции фенола (галогенирование, нитрование, сульфирование). Ориентирующее влияние гидроксигруппы.
Фено́лы — органические соединения ароматического ряда, в молекулах которых гидроксильные группы связаны с атомами углерода ароматического кольца. По числу ОН-групп различают:
· одноатомные фенолы (аренолы): фенол (C6H5OH) и его гомологи;
· двухатомные фенолы (арендиолы): гидрохинон, пирокатехин, резорцин;
· трёхатомные фенолы (арентриолы): пирогаллол, флороглюцин, гидроксигидрохинон и т. д.

Кислотные свойства
1. Диссоциация в водных растворах с образованием фенолят-ионов и ионов водорода;
2. Взаимодействие со щелочами с образованием фенолятов (отличие от спиртов);
3. Взаимодействие с активными металлами с образованием фенолятов (образующиеся в результате реакций 2 и 3) феноляты легко разлагаются при действии кислот. Даже такая слабая кислота, как угольная, вытесняет фенол из фенолятов, следовательно, фенол — ещё более слабая кислота, чем угольная).
При взаимодействии фенолятов с галогенпроизводными образуются простые и сложные эфиры (реакция Фриделя — Крафтса).


| Взаимное влияние гидроксигрупп в многоатомных спиртах. |
Так как в глицерине три группы ОН, в отличие от одноатомных спиртов, химическая активность глицерина выше, чем одноатомных спиртов. В реакциях могут участвовать одна, две или все три гидроксигруппы. Так, при взаимодействии с галогеноводородами они могут замещаться последовательно, и в конечном итоге образуется тригалогенопроизводное:
 Глицерин взаимодействует с гидроксидами некоторых металлов, в том числе и меди (II), с образованием глицератов. Эта реакция используется для обнаружения многоатомных спиртов:
Глицерин взаимодействует с гидроксидами некоторых металлов, в том числе и меди (II), с образованием глицератов. Эта реакция используется для обнаружения многоатомных спиртов:


|