СИНТЕЗ БРОМАТОВ РЕДКОЗЕМЕЛЬНЫХ ЭЛЕМЕНТОВ
(Курсовая работа)
| Выполнил Студент 1 курса, 822 группы. Земляков Д.И. |
| Научный руководитель К.х.н., доцент Батырева В.А. |
Томск 2003
СОДЕРЖАНИЕ.
ТЕОРЕТИЧЕСКИЕ ОСНОВЫНЕОРГАНИЧЕСКОГО СИНТЕЗА 3
ИОННЫЙ ОБМЕН.................................................................. 4
1.Периодическая система и её закономерности как методологическая основа неорганического синтеза................................................................... 9
2.Термодинамический анализ реакций синтеза..................... 9
3.Кинетический анализ реакций синтеза.............................. 11
3. Кинетика и механизм неорганических реакций............... 16
4.Основные методы получения веществ металлов и неметаллов. 20
5.Синтез броматов РЗЭ......................................................... 22
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ...................................... 23
СПИСОК ЛИТЕРАТУРЫ..................................................... 24
ТЕОРЕТИЧЕСКИЕ ОСНОВЫНЕОРГАНИЧЕСКОГО СИНТЕЗА
Можно выделить три аспекта синтеза: получение известных веществ по известным методикам, получение известных веществ с определенной заданной морфологией (высокодисперсных порошков, монокристаллов, тонких пленок и др.) и получение новых, ранее неизвестных веществ. В учебном практикуме на начальном этапе реальна постановка задачи синтеза известных веществ по известным методикам, и лишь в самых общих чертах возможно ознакомление с проблемой направленного синтеза веществ.
Теоретические основы неорганического синтеза в данном пособии рассматриваются применительно к задачам учебного практикума на базе знаний, полученных студентами при изучении неорганической химии. Это определяет круг включенных в данную главу вопросов и уровень их изложения. Так, в ней рассматриваются методы синтеза, доступные для учебного практикума, и не рассматриваются методы, которые, будучи даже весьма перспективными, в практикуме трудно реализуются, например синтез при высоком давлении, плазмохимический синтез. В главе не приводятся термодинамическая и кинетическая характеристики используемых реакций, за исключением самых общих соображений об их термодинамической возможности и скорости.
Методы неорганического синтеза можно систематизировать, используя разные подходы: по классам синтезируемых соединений (синтез оксидов, гидроксидов, гидридов и т.д.), по типам химических реакций, используемых в синтезе (хлорирование, гидролиз, термолиз и др.), по агрегатному состоянию реагентов (синтез в газовой, твердой, жидкой фазе), по характеру используемой аппаратуры (синтез в вакууме, низкотемпературный синтез и т.д.), по количеству используемых реагентов (макро-, полумикро-, микросинтез). Однако ни одна из этих классификаций не охватывает все разнообразие методов. Например, оксиды металлов чаще всего получают при высокой температуре, а комплексные соединения - в водном растворе. Но эти соединения можно получить и при других условиях, используя самые разные реакции. Так, для получения оксидов металлов можно использовать реакции химического или электрохимического окисления металлов в водном или неводном растворе, окисления их низших оксидов при комнатной температуре и др. При этом синтез можно вести на воздухе и в вакууме, получать вещество в микро-или макроколичестве.
Применительно к задачам практикума по неорганической химии имеет смысл уяснить особенности методов синтеза неорганических соединений разных классов, т.к. это отвечает логике построения теоретического курса. Но согласно логике построения самого практикума, когда ставится задача не только ознакомиться с методами синтеза, но и освоить некоторый объем химического эксперимента, приобрести навыки и умения выполнения определенных химических операций, особый интерес представляет возможность уяснить особенности методов синтеза в разных условиях их проведения. Как обязательная предполагается характеристика особенностей используемых в синтезе химических реакций.
ИОННЫЙ ОБМЕН
Методы ионного обмена в различных модификациях нашли в настоящее время широчайшее применение не только для аналитических целей, но и в препаративных работах неорганического синтеза. Несмотря на многообразие методов, с применением ионного обмена (распределительная хроматография, хроматография на бумаге, использование жидких ионообменников, тонкослойная хроматография и т. д.) ведущая роль по-прежнему остается за классическими методами ионного обмена.
Успешное решение любой конкретной задачи с применением метода ионного обмена зависит от правильного выбора сорбента и условий его использования. Для этого весьма существенно представлять себе структуру и свойства сорбента как:
химического соединения, так как ионообменная способность, механические и физико-химические свойства сорбентов тесно связаны с их структурой и условиями синтеза.
Ионитами называются органические или неорганические вещества, практически нерастворимые в воде или других растворителях, содержащие активные (ионогенные) группы с подвижными нонами и способные обменивать эти ионы на ионы других электролитов (поглощаемые ионы).
В зависимости от характера введения ионообменных групп все сорбенты делятся на три основных класса:
1. Сорбенты, содержащие в своей структуре кислотные группы, т. е. сорбенты, обладающие свойствами кислот и способные к обмену катионов (катиониты).
2. Сорбенты, содержащие в структуре основные группы, т. е: сорбенты, обладающие свойствами оснований и способные к обмену анионов (аниониты).
3. Амфотерные иониты, т. е. иониты, ионогенная группа которых может вести себя как кислотная или как основная, в зависимости от рН среды.
Существуют также смешанные иониты, т е. сорбенты,. в структуры которых одновременно входят как кислотные, так и основные группы.
Основные требования, предъявляемые к ионообменным смолам, следующие: высокая механическая прочность; химическая устойчивость; минимальная растворимость и небольшая набухаемость при контакте с раствором; высокая обменная способность (смола должна содержать достаточное количество пространственно доступных ионообменных групп); достаточная скорость обмена; желательная избирательность поглощения определенного типа ионов.
Катиониты могут содержать в своем составе различные кислотные группы: сульфогруппу, фосфорнокислые, карбоксильные, фенольные, мышьяково- и селеновокислые и др.
В состав анионитов в качестве функциональных групп могут входить первичные, вторичные и третичные аминогруппы, четвертичные аммониевые и пиридиновые основания.
В зависимости от величины константы диссоциации катионитов в Н+ -форме и анионитов в ОН- -форме все смолы делятся на сильно- и слабокислотные катиониты и соответственно сильно и слабоосновные аннониты.
При выборе сорбентов в первую очередь нужно учесть, с чем удобнее работать –с катионитом или анионитом. Многие задачи могут быть успешно решены и на том, и на другом типе сорбентов. Например, для разделения ионов металлов можно с успехом применить катиониты. Однако применение для этой же цели анионитов, основанное на разделении анионных комплексов этих металлов, часто бывает проще и быстрее.
Необходимо учитывать также избирательность поглощения сорбентами тех или иных ионов, которая обусловлена химической природой сорбента и определяется относительной прочностью связей обменивающихся ионов в фазе смолы. При этом энергия связи сорбируемого иона зависит не только от прочности связи этого иона с активной труппой сорбента, но и от прочности его связей с любыми другими, так называемыми неактивньгми, структурными группами ионита.
Сильные катиониты и аниониты, например, сульфокатиониты и аниониты типа четвертичных аммониевых оснований, не проявляют большой избирательности в отношении большинства ионов. Большая емкость смол такого типа, а также их способность функционировать в широком интервале рН могут быть использованы для концентрирования сильно разбавленных растворов, для обессоливания и в других случаях, когда необходимо полное извлечение всех катионов или анионов из раствора. Для выделения какого-либо одного элемента из смеси элементов бывает удобно подобрать такой сорбент, который избирательно поглощал бы ионы интересующего элемента.
В настоящее время известно большое количество селективных сорбентов. Синтез таких сорбентов сводится к задаче получения смолы с такой структурой, которая подобна структуре веществ, образующих прочные комплексы или нерастворимые соединения с данным ионом. Так была синтезирована смола (селективно сортирующая никель) путем введения в структуру смолы глиоксимовых группировок.
После выбора соответствующего сорбента необходимо определить область кислотности, в которой работает выбранный ионообменник, и его химическую устойчивость по отношению к тем рабочим средам и температурам, при которых должна проводиться очистка.
Процесс обмена ионов описывается уравнением изотермы, предложенным Б.П.Никольским:

где X1(2) и X2(2) -концентрации обменивающихся ионов в ионите (ммоль/г); X1(1)и X2(1) -концентрация обменивающихся ионов в равновесном растворе (ммоль/мл); γ1(2)и γ2(2) -коэффициенты активности обменивающихся ионов в фазе смолы; γ1(1) и γ2(1)- коэффициенты активности обменивающихся ионов в фазе раствора; Z1 и Z2 - заряды обменивающихся ионов; K -константа обмена.
Определение коэффициента активности в фазе смолы задача весьма сложная. Однако можно принять, что отношение коэффициентов активности ионов в поглощенном состоянии остается постоянным, и эту величину (можно ввести в константу. тогда, константа ионного обмена:

где a1(1) и а2(1) -активности обменивающихся ионов в равновесном растворе.
Если принять, что интересующий ион отмечен индексом 1, то для этого иона из уравнения никольскова имеем:

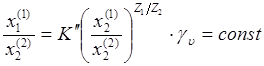
не  -множитель, содержащий отношение коэффициентов активности обменивающихся ионов в растворе (в соответствующих степенях);
-множитель, содержащий отношение коэффициентов активности обменивающихся ионов в растворе (в соответствующих степенях); 
Отношение количества вещества, поглощенного одним грамом сухой смолы, к его концентрации в равновесном растворе называется коэффициентам распределения данного иона. На практике для характеристики поглощения часто определяют именно эту величину, а не константу обмена, которая требует учёта не всегда известных коэффициентов активности.
По определению коэффициент распределения (α)

где q1(2) и q1(1) -содержание исследуемого иона соответственно фазе смолы и в растворе при равновесии, выраженное в любых единицах; V-объем раствора; m-навеска ионнта.
Коэффициент распределения являетcя величиной постоянной не зависит от концентрации интересующего иона в определенном интервале концентраций. Последнее означает, что поглощение ионов элемента -примеси прямо пропорционально его концентрации в растворе, и, следовательно, при очень малых концентрациях изотерма сорбции линейна. Величина коэффициента распределения зависит от природы второго обменивающегося иона, присутствия в растворе других ионов, в том числе мплексообразователей, кислотности раствора, температуры и давления.
Отношение коэффициентов распределения двух различных ионов в одних и тех же условиях называется коэффициентом разделения этих ионов в данных условиях.
Очистку соединений с помощью ионного обмена можно осуществлять разными способами. Если вещества-примеси содержат ионы с величиной заряда, отличающейся от величины заряда очищаемого элемента, то отделение основывается на том, что многозарядные ионы из разбавленных и умеренно концентрированных растворов поглощаются намного сильнее, чем ионы с меньшей величиной заряда. Так, если на сульфокатионитетипа КУ -2 или дауэкс -50 поглотить смесь щелочных, щелочноземельных и редкоземельных элементов, то при элюировании разбавленными растворами хлорной или соляной кислоты в первую очередь будут вымываться ионы щелочных металлов.
В более сложных случаях, когда необходимо разделить элементы, коэффициенты разделения которых близки к единице, чаще всего используют метод комплексообразовательной хроматографии. В этом случае весьма существенными становятся данные о составе, условиях образования и константах устойчивости различных комплексов разделяемых элементов.
В первом варианте все разделяемые ионы сначала поглощаются смолой. Затем производят их (разделение, пропуская (через колонку со смолой раствор комплексообразоаателя., который раздвигает -первоначально образовавшиеся близко расположенные зоны и последовательно вымывает их. При этом подбирают условия, наиболее благоприятные для комплексообравования (рН, температура, концентрация, скорость.пропускания раствора и т. п.). Все ионы вымываются в строго определенном порядке, который задается соотношением прочности связи данного иона со смолой с прочностью образующихся в фильтрате комплексов. Первыми вымываются те ионы, которые образуют наиболее прочные комплексы и слабее всего удержшваютоя смолой.
Во втором варианте комплексообразователь, добавляют к раствору, содержащему смесь разделяемых элементов, и в этом растворе создают условия, благоприятствующие комплексообразованию. Затем производят сорбцию этой смеси комплексов на соответствующем ионите, например, на анионите, если были получены анионные комплексы. При этом лучше всего сорбируютоя наиболее прочные комплексы, которые имеют наибольшее сродство к смоле.
Чем больше различие констант устойчивости, использованных для разделения комплексов, тем полнее и эффективнее достигаемое разделение в обоих вариантах.
Знание констант устойчивости различного вида комплексов очень полезно также при выборе сорбентов, селективно поглощающих определенные ионы. Известно, что во многих случаях сорбированные ионы образуют комплексные соединения со структурными элементами смолы. Очевидно, что чем более
прочные комплексы образуются в фазе смолы, тем большей (избирательностью в отношении данного иона будет обладать смола. В литературе имеется немного работ, посвященных изучению прочности комплексов с функциональными группами:молы. Поэтому, на практике при выборе селективного сорбента пользуются данными об устойчивости аналогичных комплексов в растворах.
Комплексообразование в фазе смолы объясняет, например, высокую избирательность карбоксильных и фосфатных катионов в отношении некоторых катионов. Установлен следующий порядок селективности фосфорнокислых смол в отношении катионов: Th4+ >U 4+ >UO22+  Fe3+> редкоземельные элементы > Н+ > Сu2+ >Со2+ >Вa2+ >Na+.
Fe3+> редкоземельные элементы > Н+ > Сu2+ >Со2+ >Вa2+ >Na+.
Известно также, что многие ионы образуют весьма прочные хелатные комплексы. Оказалось, что смолы, синтезированные (а основе хелатообразующих соединений, обладают весьма высокой избирательностью
по отношению к катионам различных металлов. Поведение хелатных ионитов во многих отношениях сходно с поведением обычных хелатных соединений. В частноси, образование хелатов в фазе ионита сильно зависит от рН и поглощение увеличивается с ростом рН раствора.
Аниониты также обладают способностью координационно связывать некоторые катионы, имеющие ярко выраженную тенденцию к образованию анионных комплексов.
1.Периодическая система и её закономерности как методологическая основа неорганического синтеза.
Прежде всего, методологической основой неорганического синтеза являются периодический закон и периодическая система с ее закономерностями (правило об уменьшении стабильности высшей степени окисления с ростом атомного номера в главных подгруппах, диагональное сходство, близость атомных радиусов у атомов элементов пятого и шестого периода за счет f-сжатия, способность элементов к диспропорционированию, полимеризации, комплексообразованию и др.), теории кислотно -основных реакций, теории сольволиза и гидратации, учение о механизмах химических реакций (окислительно-восстановительных, радикальных, обмена лигандов и т.д.), теории химической связи, основные законы химии (при синтезах, например, закон эквивалентов дополняется положением о возможности для многоосновных кислот, многовалентных атомов элементов существования переменного значения кислотно -основного, окислительно-восстановительного эквивалентов).
Термодинамический анализ реакций синтеза.
Итак, при термодинамической оценке пригодности для синтеза какой-либо обратимой реакции
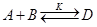
необходимо, чтобы было отрицательным изменение энергии Гиббса реакции

где

Если для некоторой реакции К >1, то реакцию можно считать практически необратимой. При значении К >1 ожидается достаточно большой выход продуктов, при К <1 реакция должна протекать в основном "справа налево". Условие К <1 не означает, что реакция синтеза продукта не совершается, в этом случае необходимо вычислять равновесный выход продукта, хотя он может быть и мал.
Уравнение, связывающее изменение энергии Гиббса реакции с константой равновесия

можно записать в виде

Подставив в это уравнение величины R = 8,З1 ·10-3 кДж/К ·моль и Т= 298 К, получим
∆G°298 = -2,3 8,31 ·10-3· 298 ·ln K298 = -5,70 ln K298.
При значении K ~107 реакция практически проходит до конца в прямом направлении, поэтому значение ∆G° ~ |40| кДж/моль (5,7ּln107 = 5,7ּ7 ~ 40) можно считать в первом приближении границей возможности или невозможности самопроизвольного протекания реакции. Если ∆G° реакции при данной температуре отрицательно и по модулю больше, чем 40 кДж/моль, то такая реакция может протекать в прямом направлении не только при стандартных, но и при любых других условиях. Если же ∆G° реакции при данной температуре положительно и больше 40 кДж/моль, то такая реакция протекать самопроизвольно ни при каких условиях не может.
Если значение ∆G° реакции по абсолютному значению невелико, то при изменении условий процесс может протекать в том или ином направлении. В этом случае для решения вопроса о направлении самопроизвольного протекания реакции недостаточно определить знак и величину ∆G°, нужно рассчитать значение ∆G○ реакции с учетом содержания во взятой смеси исходных и образующихся веществ.
С помощью известных констант равновесия химических процессов можно решить два вопроса: 1) предсказать направление самопроизвольной реакции при заданных условиях эксперимента и 2) при известных исходном составе системы и константе равновесия можно рассчитать равновесный состав смеси, максимально возможный "выход" продуктов, что важно для реакций синтеза. Рассмотрим это на примере расчета газовых реакций.
При решении первого вопроса удобно ввести понятие кажущейся константы равновесия Q как отношения концентраций продуктов к концентрациям реагентов, но не обязательно относящееся к равновесным условиям. Для нахождения Q можно взять отношение, например, начальных концентраций компонентов (C°). Так, для реакции

 ,
, 
Если имеется больше молекул исходных реагентов, чем должно быть при равновесии, то увеличение знаменателя в выражении для Q приводит к тому, что Q <K, и реакция самопроизвольно пойдет в прямом направлении для образования большего количества продукта; при Q >К самопроизвольно протекает обратная реакция, а при Q =К реагенты и продукты находятся в равновесии.
Определение выхода продуктов реакции проводят с использованием таких понятий, как степень диссоциации α, степень превращения γ, число прореагировавших молей ξ.
Степенью диссоциации α называют долю газа, распавшегося на продукты к данному моменту времени. Значение α можно вычислить для известного значения константы равновесия.
Выразив α через Кр (константа равновесия) и давление компонентов P, получим:

Выражение степени диссоциации компонентов раствора.