Предметом химической кинетики являются скорости реакций со всеми влияющими на них факторами и интерпретация скорости реакций на основе их механизма. В этом смысле кинетика отличается от термодинамики, в которой рассматриваются начальное и конечное состояния системы вне зависимости от времени протекания этого превращения. Термодинамика обычно рассматривает системы в состоянии равновесия, т. е. в состоянии, в котором скорости прямой и обратной реакций в обратимом процессе равны, что связывает эти две области химии. Однако обратное не верно: скорость реакции нельзя определить только на основе термодинамических данных. Химическую кинетику можно считать более фундаментальной областью науки, но, к сожалению, часто сложность исследуемых процессов делает применение теории химической кинетики довольно трудным.
Рассмотрим равновесие водород -йод в газовой фазе при высокой температуре:
Н2 + I2 = 2НI
Эта реакция протекает при бимолекулярных столкновениях между молекулами йода и водорода, так что стехиометрическое уравнение в этом случае соответствует истинному механизму взаимодействия. Доказательством правильности такого механизма является то, что скорость образования йодистого водорода, как было показано, пропорциональна как концентрации водорода, так и концентрации йода, т. е.

Это, однако, исключительный случай, так как обычно стехиометрическое уравнение не описывает механизма реакции, как, например, для значительно более сложной реакции водорода с бромом, скорость которой, как было показано, определяется уравнением:

Это можно объяснить следующим механизмом:

Br + Н2 → НВr + Н (медленная)
Н + Вr2 → НВr + Вr (быстрая)
Н + НВr → Н2 + Вr (быстрая)
Следует отметить, что, зная механизм реакции, не всегда можно дать достаточно определенную интерпретацию экспериментально найденным выражениям для скорости. Иногда с экспериментальными данными согласуются несколько возможных механизмов или вновь полученные данные опровергают ранее принятый механизм.
Очевидно, математическую обработку выражений скоростей реакций через концентрации в определенных степенях
Уравнение:1
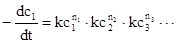
проводить легче, чем для выражений более сложного типа. Только для выражений скорости типа уравнения (1) приемлемо определение порядка реакции n, причем
Уравнение:2
n = n1+n2+n3+……
Из двух рассмотренных выше примеров реакция водорода с йодом -это реакция второго порядка, причем как по водороду, так и по йоду порядок ее равен единице. Понятие порядка реакции неприменимо к взаимодействию водорода с бромом, так как выражение для скорости этого процесса записано не в соответствующей форме.
Если условия проведения реакции таковы, что одна или более концентраций остаются практически постоянными в течение опыта, то эти концентрации можно включить в константу скорости k. В этом случае реакция будет иметь псевдо –n порядок, где n -сумма показателей степеней концентраций, которые в течение эксперимента изменяются. Обычно эти показатели степени -простые положительные числа, но в зависимости от сложности реакции они могут быть дробными или даже отрицательными.
Порядок реакции, определяемый уравнением (2), часто путают с молекулярностью реакции, которая определяется числом молекул, участвующих в элементарном процессе столкновения. Таким образом, молекулярность - это теоретическое понятие, проистекающее из принятого механизма реакции, тогда как порядок - величина эмпирическая; эти две величины могут различаться. Однако бимолекулярные реакции обычно имеют второй порядок, а тримолекулярные реакции -третий порядок, но обратное утверждение не всегда верно. Реакция, которая иллюстрирует только что сказанное, -это окисление ионов Fe2+ перекисью водорода. Стехиометрическое уравнение ее выглядит так:
2Fr2+· aq + Н2О2 → 2Fe3+ · aq + 2OH-
Показано, что выражение для скорости этой реакции

т. е. реакция имеет второй порядок. Схему протекания реакции лучше всего можно представить следующими стадиями:
Fe2+· aq + Н2О2  Fe3+ • ао+ОН" +ОН
Fe3+ • ао+ОН" +ОН
и 
Fe2+ · aq + OH-  Fe3+· aq + OH-
Fe3+· aq + OH-
где
k1= 60 л/моль · сек и k2 = 60 000 л/моль · сек
Так как суммарная реакция состоит из двух последовательных бимолекулярных стадий, то какую -либо молекулярность стехиометрическому уравнению приписать нельзя. Эта схема также иллюстрирует тот факт, что скорость всего процесса определяет самая медленная стадия, так как константа скорости суммарного процесса - это константа скорости первой, более медленной бимолекулярной стадии (т. е. k = k1). Вторую стадию в этой схеме можно использовать как пример реакции с псевдопорядком.
Для объяснения экспериментальных данных по механизмам реакций широко используют явление изотопного замещения. Так, образец, содержащий радиоактивные ионы Fe2+, можно обработать нерадиоактивным образцом, содержащим ионы Fe3+, и количество полученных радиоактивных ионов Fe3+ можно измерить в зависимости от времени. Уравнение Маккея

связывает скорость реакции R (т.е. скорость обмена радиоактивностью) с начальными концентрациями a и b реагентов и измеренными радиоактивностями х и  первоначально неактивной формы (в данном случае Fe3+) в моменты времени t и
первоначально неактивной формы (в данном случае Fe3+) в моменты времени t и  . Поэтому такие реакции являются идеальными для исследования влияния температуры, концентрации и других факторов на скорость реакции.
. Поэтому такие реакции являются идеальными для исследования влияния температуры, концентрации и других факторов на скорость реакции.
Таким образом, истинный механизм химических реакций включает мономолекулярные, бимолекулярные или тримолекулярные стадии, по которым реакция идет самопроизвольно при столкновениях между двумя или тремя молекулами. Вероятность одновременного столкновения четырех или более молекул настолько мала, что ею можно пренебречь. Однако можно легко показать, что не все столкновения приводят к химическому взаимодействию. Основными ограничениями, которые лимитируют эффективность столкновений, являются:
а) ориентационные эффекты; очевидно, сложные молекулы могут вступать в реакцию только тогда, когда они соударяются в определенных положениях и в соприкосновение приходят реакционноспособные связи или неподеленные пары электронов. Стерический фактор p показывает, какая часть общего числа соударений приходится на столкновения молекул с такой ориентацией;
б) энергия активации; рассмотрим простую реакцию в газовой фазе
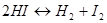
Расстояние H -I в молекуле йодистого водорода равно 1,61 Å и диаметр молекулы равен 3,5 Å.Этот диаметр также должен быть равен расстоянию между двумя атомами водорода или двумя атомами йода в соударяющихся молекулах (удвоенный вандерваальсов радиус; разд. 4.2). Естественно, это расстояние велико по сравнению с расстояниями в молекулах водорода (0,74 Å) и йода (2,67 Å). Следовательно, соударения должны обладать достаточной энергией, чтобы вызвать сжатие молекул НШ, после чего составляющие атомы имели бы возможность подойти друг к другу достаточно близко и вызвать распад этих молекул на водород и йод. Необходимую для этого энергию называют энергией активации реакции, и только те столкновения, которые имеют это минимальное количество энергии, будут эффективными. Часть таких столкновений определяется выражением  , где Еa -энергия активации столкновений на один моль. Константа скорости определяется уравнением Аррениуса
, где Еa -энергия активации столкновений на один моль. Константа скорости определяется уравнением Аррениуса

где Z - общее число столкновений между реагирующими молекулами в единичном объеме за одну секунду. Такое простое изложение теории соударений показывает, что она основана на кинетической теории.
Предполагают, что когда взаимодействуют две молекулы, обладающие необходимой энергией активации, то они вначале образуют активированный комплекс, или переходное состояние, который затем разлагается с конечной скоростью с образованием продуктов реакции. Принимают, что скорость реакции определяется скоростью прохождения через переходное состояние, т. е. скоростью прохождения через потенциальный энергетический барьер.Концентрация активированного комплекса в любой момент определяется его равновесием с исходными молекулами. Высота барьера по отношению к энергии исходного состояния равна энергии активации, а разность между энергиями начального и конечного состояний равна теплоте реакции.
3. Кинетика и механизм неорганических реакций.
С кинетической точки зрения неорганические реакции можно подразделить на две группы:
а) реакции, включающие разрыв и образование ковалентных связей, и
б) реакции, сопровождающиеся простым переносом электронов,
Кроме того, в твердом состоянии реакции протекают еще при перемещении ионов из одной решетки в другую по дефектам решетки. Первый класс реакций можно подразделить на реакции, подобные термическому разложению, рассмотренному ранее, и реакции замещения в координационных соединениях, в которых координированный лиганд замещается другим лигандом из раствора. В общем случае реакции замещения по своему характеру нуклеофильные, так как замещаемый лиганд уносит электронную пару, ранее образовывавшую 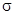 -связь металл -лиганд, а замещающий лиганд приносит пару электронов и поэтому занимает положение с низкой электронной плотностью. По аналогии с органическими соединениями эти процессы обозначаются как SN-процессы (нуклеофильное замещение). Возможны два основных пути протекания реакции в зависимости от того, происходит ли предварительная диссоциация реагирующего комплекса (мономолекулярный процесс SN1)
-связь металл -лиганд, а замещающий лиганд приносит пару электронов и поэтому занимает положение с низкой электронной плотностью. По аналогии с органическими соединениями эти процессы обозначаются как SN-процессы (нуклеофильное замещение). Возможны два основных пути протекания реакции в зависимости от того, происходит ли предварительная диссоциация реагирующего комплекса (мономолекулярный процесс SN1)

или важной стадией является бимолекулярный процесс замещения, скорость которого зависит от концентрации как комплекса, так и замещающего лиганда (SN2), т. е.

Следовательно, SN1 -механизм должен привести к активированному комплексу, в котором ион металла имеет меньшее координационное число, чем в исходном комплексе, тогда как SN2 -механизм требует увеличения числа присоединенных лигандов в переходном состоянии. Необходимо далее рассмотреть разность энергии между реагирующим комплексом и этими переходными состояниями. Если в комплексе нет 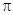 -связей металл -лиганд, то величину скорости реакции можно предсказать, предполагая электростатическое взаимодействие между ионами металла и лигандами.
-связей металл -лиганд, то величину скорости реакции можно предсказать, предполагая электростатическое взаимодействие между ионами металла и лигандами.
Наличие двух «вакантных» гране -положении в комплексе с конфигурацией плоского квадрата позволяет предположить, что в этом случае более вероятен SN2-механизм. В действительности, однако, почти наверное эти транс- положения не будут свободными, и если нет других лигандов, то они будут заняты молекулами растворителя. Эти молекулы растворителя находятся на большем расстоянии, чем лиганды в плоскости квадрата. Поэтому комплекс будет вести себя во многих отношениях так, как если бы он имел конфигурацию плоского квадрата. Две координированные молекулы растворителя очень подвижны и легко могут быть замещены лигандами из раствора. Это облегчает замещение наиболее подвижного лиганда в плоскости квадрата, например
 |
Здесь S -молекула растворителя, а рисунок не представляет собой никакой частной стереохимической конфигурации пятикоординационного переходного состояния. К этому переходному состоянию легко присоединяется нуклеофильный реагент У; одновременно комплекс теряет молекулы растворителя и образуется новый комплекс [ML3Y]. Экспериментально было найдено, что уравнение для скорости реакции типа
[ЭIIX4] + Y→ [ЭIIX3Y] + 3
имеет вид
скорость = k1 [комплекс] + k2 [комплекс] [Y]
где k1 -константа скорости реакции первого порядка, которую относят к процессу с SN2 -механизмом; в этом процессе растворитель -нуклеофильная атакующая единица, k2 -константа скорости реакции второго порядка в процессе с SN2 -механизмом, в котором нуклеофильной единицей является Y. Если расположить нуклеофильные реагенты в порядке возрастания k1 или k2 то их реакционная способность по отношению к элементу с положительной степенью окисления II будет увеличиваться в ряду:
H2O ~ OH- < Cl- < Br- ~ NH3 ~ олефин < ру < NO-2 < N-3 < I- ~ SCN- ~ PR3
Очевидно, что по отношению к платине со степенью окисления II большую реакционную способность имеют те лиганды, которые могут быть как σ -донорами, так и π -акцепторами. Платина, занимающая место в конце третьего ряда переходных элементов, имеет несвязанные электроны, необходимые для образования π -связей металл –лиганд. Приведенный выше порядок лигандов определяет также повышение реакционной способности других лигандов, находящихся по отношению к первым в транс -положении. Это явление называют транс -влиянием. Так, в реакции

для цис –изомера k1 = 1,7 ·10-2 (при 0°), а для транс -изомера k1 = 10 ·10-6 (при 25°), т. е. когда замещаемый лиганд (Cl-) находится в транс -положении по отношению к фосфиновой группе, то его замещение идет намного легче, чем когда он находится в транс -положении по отношению ко второму хлору. Можно ожидать, что в комплексах типа 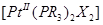 π -связь металл -лиганд прочнее для цис-, чем для транс -изомера. Два атома фосфора в транс -изомере (предполагается, что они лежат вдоль оси х молекулы) могут использовать для образования π -связи только
π -связь металл -лиганд прочнее для цис-, чем для транс -изомера. Два атома фосфора в транс -изомере (предполагается, что они лежат вдоль оси х молекулы) могут использовать для образования π -связи только  и
и  орбитали, а в цис -изомере
орбитали, а в цис -изомере  и орбитали. Разность сил связей будет максимальной, когда лиганды Х - слабые π -акцепторы, как в случае иона хлора. Когда же подвижные лиганды не могут действовать как π -акцепторы, их различие в проявлении транс -влияния должно иметь электростатическую природу. Это можно объяснить, предположив поляризуемость подвижного лиганда, влияющего на электронное распределение вокруг центрального атома: этот тип индуктивного эффекта мало проявляется в цис -положении (через 90°), но на лиганд в транс -положении подвижный лиганд оказывает наибольшее влияние. Встречаются такие случаи, когда влияние л-связи и электростатического эффекта само по себе мало, но они дополняют друг друга, приводя к довольно сильному суммарному влиянию на транс -положение; это наблюдается, например, для I-. Эти предсказания подтверждаются экспериментально исследованием спектров -ядерного магнитного резонанса и определением энергий связей. Так, общая энергия связи цис -
и орбитали. Разность сил связей будет максимальной, когда лиганды Х - слабые π -акцепторы, как в случае иона хлора. Когда же подвижные лиганды не могут действовать как π -акцепторы, их различие в проявлении транс -влияния должно иметь электростатическую природу. Это можно объяснить, предположив поляризуемость подвижного лиганда, влияющего на электронное распределение вокруг центрального атома: этот тип индуктивного эффекта мало проявляется в цис -положении (через 90°), но на лиганд в транс -положении подвижный лиганд оказывает наибольшее влияние. Встречаются такие случаи, когда влияние л-связи и электростатического эффекта само по себе мало, но они дополняют друг друга, приводя к довольно сильному суммарному влиянию на транс -положение; это наблюдается, например, для I-. Эти предсказания подтверждаются экспериментально исследованием спектров -ядерного магнитного резонанса и определением энергий связей. Так, общая энергия связи цис -  больше, чем энергия транс -изомера, примерно на 10 ккал.
больше, чем энергия транс -изомера, примерно на 10 ккал.
В тетраэдрических комплексах связи металл -лиганд обычно более подвижны, кроме случаев, когда в состав комплекса входят хелатообразующие лиганды. Например, комплекс бериллия с бензоилацетоном можно разделить на оптические антиподы методом избирательной адсорбции одной энантиоморфной формы на колонке с оптически активным кварцем.