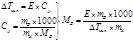Обратимый
ΔS=Qобр/T
Qобр-TΔS=0; p=const
ΔH-TΔS=0;
ΔG=0 критерий равновесного состояния системы
Необратимый
ΔS>Qнеобр/T
Qнеобр-TΔS=0;
ΔH-TΔS<0;
ΔG<0 критерий самопроизвольности необратимого процесса
Для хим реакций ΔrG˚ рассчитывается по следствию из закона Гесса
ΔrG˚=ΣконΔf*G˚*ni- ΣначΔf*G˚*ni
T=const
Обратимый
V=const; Qv=ΔU
Qобр-TΔS=0
ΔU-TΔS=0 ΔF=0
Необратимый
V=const; Qv=ΔU
Qобр-TΔS=0
ΔU-TΔS<0 ΔF<0
Для процессов, протек при T, р=const, условием самопроизвольного течения явл уменьшение энергии Гиббса. Причем условием их равновесия явл достижение минимального значения для данного условия ф-ии G.
—1 dp=0 dT=0 dG=<0
—2 dV=0 dT=0 dF=<0
В хим т/д большее значение имеет ф-ия, наз хим пот-лом (μi).
μ i – ф-ия характеризует состояние к-либо i-компонента в фазе данного состава при опр местных условиях.
Хим пот-л – приращение изобарно-изотермичесокго пот-ла данной фазы при введении дополнит кол-ва i-компонента, при T,р=const и постоянных кол-вах др компонентов, содержащихся в данной фазе.
μi=(д Gi/ д ni)p,T,j=i
G=Σμi*dni
Хим пот-л зависит от концентрации данного компонента и его вида, а также от вида и концентрации др компонентов в этой фазе.
Только в случае идеальн газа хим пот-л опр видом и концентрацией i-компонента и не зависит от концентрации др компонентов.
μi=μi˚+RTln(pini)
pi- парциальное давление i-компонента данной смеси
μi˚- хим потенциал, при парциал давлении =1
ni- кол-во вещества
Общим условием возможности самопроизвольного процесса будет равенство
Σμi*dni=0
Рассмотрим гомоген газ реакцию, к-ая протекает по заданному у-ию
υ1A1+ υ2A2↔ υ3A3+ υ4A4 n=υ
Эта обр хим реакция протекает до тех пор пока не установится равновесие между реагир в-вами. Кол-венно хим равновесие описывается з-ном действия масс.
V1=k1*p A1υ1*pA2υ2
V2=k2*p A3υ3*pA4υ4 V1=V2 условие равновесия
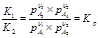
Закон действия масс
Выражение для хим потенц участников данной реакции
µ1=μ1˚+RTln(p1n1) исх в-ве
μ2=μ2˚+RTln(p2n2) ∆G=μ1+μ2
μ3=… кон в-ве
μ4=… ∆G=μ3+μ4
∆rG=∆конG-∆исхG=(μ3+μ4)-(μ1+μ2)=∆rG˚+RTln((p3υ3*p4υ4)/(p1υ1*p1υ1))
∆rG=∆rG˚+RTlnKP
в усл равнов ∆rG˚=-RTlnKp˚ ∆rG=0
условие нормального сродства или изотерма хим реакции в стандарт условиях. Задавая произвольное значение pi≠1, получаем
∆rG=-RTlnKP+ RTlnПpi где Пpi=(Пpiкон)/(Пpiисх)
↑ уравнение изотермы химич реакции
хим т/д позволяет определить константу равновесия какой-либо реакции при другой температуре, не ставя дополнительного эксперимента. Для этого существует ур-е изобары.
Ур-е изобары получается при дифференцировании уравнения изотермы химич реакции по температуре, и комбинировании полученного ур-я с ур-ем Гиббса-Гельмгольца
ΔG=-RTln(K˚)+RТln(Пpi)
ΔG=ΔH-T(д ΔG/ д Т)
dln(Kp)/dT=ΔH/(RT2) изобара
dln(Kс)/dT=ΔU/(RT2) изохора
Чтобы получить удобное для расчета мат выражение для ур-я изобары следует проинтегрировать дифференцированную форм; предполагая, что в небольшом интервале температур Т1–Т2 тепловой эффект реакции от температуры не зависит. Тогда получаем интегральную форму
ln(K1/K2)=ΔH(T1-T2)/(R*T1*T2) интегральная форма изобары
Следствия:
1) Можно рассчитать константу равновесия при Т2, отличающуюся от Т1 если известна К1 и тепловой эффект реакции в этом интервале темпер
2) Можно рассчитать тепловой эффект реакции если известны константы равновесия хотя бы при двух температурах
3) С помощью уравнения подтверждается вывод и- принципа смещения равновесия Ле-Шателье (влияние на константу равновесия температуры)
Если на систему, находящуюся в равновесии оказывать внешнее воздействие, то равновесие сместится в сторону реакции, противодействующей внешнему влиянию
3H2+N2=2NH3-ΔH
(г) (г) (г)
- увеличение P → прямая
- увеличение Т → обратная
ln(K1/K2)=(ΔH/R)*(T1-T2)/(T1*T2)
Химическая кинетика
Кинетика хим реакций– учение о скорости их протекания и зависимости ее от различных факторов. Такие факторы: строение молекулы, концентрация участников реакции, температура, свойства среды, наличие катализатора, внешние воздействия.
По правилам ИЮПАК скорость химической реакции определяется как возрастание степени завершенности реакции V=dξ/dτ
Более удобно понятие скорости образованя или преварщения некоторого компонента в системе Vi=±dCi/dτ
Влияние концентрации на скорость описывается законом действующих масс
Скорость реакции пропорционально произведению концентраций веществ, взятых в степенях, равных стехиометрическим коэффициентам
V=K*C1n1*C2n2*…*Cini
Вид уравнения зависит от того, как протекает реакция
А+2В=С V=K*CА1*CВ2
N2+3H2=2NH3
V=K*CN2*CH23
К– коэффициент пропорциональности химических реакций
Физический смысл константы скорости – скорость химической реакции при единичной концентрации компонента. Сумма показателей степеней ΣNi=n: общий порядок реакции
Показатель ni называется порядком реакции по i-тому компоненту
В простых реакциях в одну стадию ni– целое число и его значение совпадает с молекулярностью реакции
Молекулярность определяется числом молекул в элементарном хим акте (целое положительное число (1,2,3)); элементарных актов с учатием 4-х молекул не бывает
Влияние темперауры
При повышении Т процесса на каждые 10˚ скорость реакции в области умеренных температур увеличивается 2-4 (Вант-Гофф)
V2/V1=K2/K1=γ — отношение констант скорости (γ=2–4) реакций при 2-х температурах, отличающихся на 10˚ V2=V1*γ(T2-T1)/10
А+В=С
Т1→V1=K1*CA*CB
Т2→V2=K2*CA*CB
Аррениус показал, что К зависит от температуры и эта зависимость описывается: dln(K)/dT=Еакт/(RT2) Еакт– энергия активации
Предполагая, что Еакт не зависит от природы веществ проинтегрируем данное уравнение К=Ае-(Еакт/RT) A– предэкспотенциальый множитель
Для расчета Еакт и А проводят экспериментальное определение констант скоростей, при некоторых температурах и строят график зависимости lnK=f(1/T)
Графический способ определения энергии активации.
Энергетический способ определения энергии активации.
Записывая ур-е Аррениуса для 2-х температур, но для одной реакции; делят ур-е почленно при этом сокращается А; частное,полученное от деления логарифмируем и преобразуем в формулу с выделением Еакт.
Физический смысл энергии активации
Аррениус высказал, что молекулы вступают во взаимодействие при столкновении друг с другом (разлетаются, сцепляются). Не все столкновения результативны, эффективны только столкновения между молекулами, обладающими некоторым избытком внутренней энергии по сравнению со средним значением для данной температуры. Этот избыток энергии Аррениус назвал энергией активации.
Гипотеза Аррениуса легла в основу теории активных соударений. Согласно этой теории в равнении Аррениуса А= общему числу столкновений за единицу времени. Ае-(Еакт/RT)– число активных соударений. Теория активных соударений хорошо описывает уравнения в газовой фазе. Для реакций в жидкой фазе была разработана теория переходного состояния. Основное положение теориии: при элементарном акте химической реакции образуется промежуточный активный комплекс с неустойчивыми связями и существующий очень короткое время. В результате акта комплекса с другими молекулами происходит разрушение валентных связей и комплекс распадается с образованием продуктов реакций (А+В)↔(А…В) (А…В)↔(А+В)
Кинетические уравнения
-dc/dt=k*Cn –порядок реакции®кинетическое уравнение в дифференц форме
Реакции (1) порядка (n=1)
Со – нач концентрация реагир в-ва
С - текущая концентрация
-dc/dt=k*C
Со∫С-dC/C=0∫tkdt
ln(C0/C)=k*t кин ур-е I порядка в интегр форме
Определение константы скорости реакции (1) порядка:
1) метод подстановок
В разные моменты времени опр концентрация реагир в-ва, подставляют эту концентрацию в у-ие и соответственно рассчитывают константу скорости
2) графический метод
3) по времени полупревращения
Временем полупревращения реакции (t1/2) наз время, за к-ое концентрация реагирующего в-ва, равная вначале Со, уменьшается вдвое, т.е. до1/2* Со
K=(2.303/ t1/2)*lg(Со/(1/2* Со))= (2.303/ t1/2)*lg2=0.693/t1/2
t1/2=0.693/k
Для любой конкретной реакции (1) порядка время полупревращения явл константой и не зависит от нач концентрации.
Реакции (2) порядка (n=2)
Кинетич у-ию (2) порядка подчиняется реакция следующего типа:
1)- 2A→прод реакц
2)- A+B→прод реакц CA=CB
-dc/dt=k*C2
Со∫С-dC/ C2=0∫tkdt
-1/C0+1/C=kt 1/C=1/C0+kt кин ур-е II порядка в интегр форме
k=1/t*((C0-C)/(C0*C))
Определение константы скорости реакции (2) порядка:
1) метод подстановок
В разные моменты времени находим текущую концентрацию, подставляют эту концентрацию в у-ие и соответственно рассчитывают константу скорости
2) графический метод
3) по времени полупревращения
Для реакции (2) порядка время полупревращения обр-пропорц нач концентрации
k=1/t1/2*((C0-(1/2)*C)/(C0*(1/2)*C))
k=1/t1/2*(1/C0) → t1/2=1/(k*C0)
Для реакций II порядка время t1/2 обратно пропор начальной концентр (C0)1 и (C0)2
(t1/2)1/(t1/2)2=(C0)2/(C0)1 реакция II порядка
Катализ и катализаторы
Катализ – явление селективного изменения скорости хим реакции по средствам катализатора.
Катализатор – в-во, изменяющ скорость хим реакции, участвующее в ней, но к моменту образования продуктов реакции кол-во и состав катализатора остается неизменным.
Особым видом явл автокатализ – скорость хим реакции изменяется под действием продуктов реакции.
Различ катализаторы:
- гомогенные (находятся с реагир в-вами в одной фазе)
- гетерогенные (находятся в другой фазе, чем реагир в-ва)
Они также бывают:
- положительные (увелич скорости хим реакции)
- отрицательные – ингибиторы (уменьш скорости хим реакции)
Хар-ки кат-ра:
а) активность или производительность A-активность
А=mпрод/(t*mкат) А=mпрод/(t*Sкат) А=mпрод/(t*Vкат)
б) селективность или избирательность
HC≡CH→(H2)→ H2C=C H2→(H2)→ H3C-CH3
C2H5OH →Al2O3→ C2H4+H2O
C2H5OH →Ag, Cu→ CH3CHO+H2
S(селективность)=mцелевого продукта/mвсех продуктов
Общие закономерности катализа:
1) применение кат-ра не изменяет т/д кинетич реакции, т.е. вел-ны ∆H и ∆G
- кат-р ускоряет только те хим реакции, для к-ых ∆G<0
- кат-р ускоряет достижение состояния равновесия в случае обратимых хим реакций, но не смещает равновесие и не изменяет величины константы
- применение кат-ра не изменяет тепл эффект хим реакции, т.е. ∆H
2) Кат-р уменьшает полную энергию активации реакции, т.к. каталитическая реакция идет по др пути.
А+В→АВ
А+В→А … В→АВ – без катализатора
А+В+катал→А …кат-ор … В→АВ – с катализатором
Растворы
Коллегативные свойства растворов
Коллегативные свойства растворов - свойства раствора связанные друг с другом и обусловленные общими причинами, главным из которых является число растворенных частиц в растворе.
К таким свойствам относятся: понижение давления насыщенного пара растворителя над раствором, увеличение температуры кипения, уменьшение температуры кристаллизации, осмотическое давление
I закон Рауля
Относительное понижение давления насыщенного пара растворителя над раствором равно мольной доле растворимого вещества
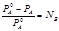 А – растворитель
А – растворитель
В – растворенное вещество
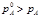 pA°–давление насыщ пара с растворителем над растворителем
pA°–давление насыщ пара с растворителем над растворителем
pA–давление насыщ пара с растворителем над раствором
Следствие
понижение давления насыщенного пара растворителя над раствором является понижением температур кристаллизации и повышения температур кипения
II закон Рауля
Депрессия кипения или кристаллизации (ΔTкр, ΔTкип) прямо пропорциональна концентрации растворенного вещества
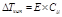
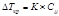
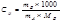
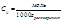
моляльная масса
Е (эбуллиоскопическая константа) характеризует свойства растворителя и численно равна депрессии кипения одномолярного раствора
EH2O=0,520 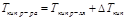
К (клиоскопическая константа) характеризует свойства растворителя и численно равна понижению температуры кристаллизации одномолярного раствора.
KH2O=1,860 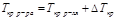
Депрессия кипения и кристаллизации величины положительные
Е и К связаны с температурой кипения и кристаллизации растворителя, а также с их удельной теплотой парообразования и плавления соответственно
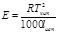

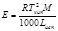
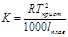
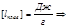
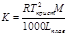
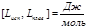
Измеряя температуру кипения и температуру кристаллизации можно определить молекулярную массу растворенного вещества