Характеристику органического соединения нельзя считать исчерпывающей, если для него приведены лишь элементный состав и молекулярная масса. Для идентификации соединения необходимо определить также другие данные, прежде всего физические свойства. Важнейшие из них — это температура кипения, температура плавления, плотность, показатель преломления, для некоторых соединений вращение плоскости поляризации света, УФ-, ИК- и ЯМР-спектры, а также распад ионизированных молекул в газовой фазе (масс-спектры).
Полученные при идентификации данные могут одновременно служить и для оценки чистоты вещества. Вещество можно признать чистым только тогда, когда физические константы его не меняются после повторной очистки — перегонки, перекристаллизации, хроматографирования и т. д.
ТЕМПЕРАТУРА ПЛАВЛЕНИЯ
Температурой плавления соединения называют температуру, при которой его твердая фаза находится в равновесии с собственными расплавом. Чистые вещества обладают четко выраженной температурой плавления; ее точное определение (±0,01 °С) возможно путем снятия кривых плавления.
При применении обычных простых методик определения температуры плавления, которые описаны ниже, плавление вещества наблюдается в интервале температур от нескольких десятых градуса до целого градуса. Незначительные загрязнения иногда сильно понижают температуру плавления данного соединения; при загрязнении вещества более высокоплавкими примесями тоже наблюдается, как правило, понижение температуры плавления. Кроме того, в этих случаях наблюдается значительное увеличение интервала плавления (более чем на 1 °С). Такое явление используется при установлении идентичности двух веществ с одинаковой температурой плавления. Для этого смешивают равные количества сравниваемых веществ (проба смешанного плавления). Если температура плавления смеси остается неизменной, то делают заключение об идентичности обоих веществ. Если же температура плавления пробы ниже температуры плавления исходных веществ, то, следовательно, имеются два разных вещества. Однако для смесей изоморфных соединений даже разных по химическому составу понижение температуры плавления не обнаруживается.
Надо выработать привычку первоначально подтверждать идентичность двух веществ, используя эти простые и быстрые методы, а уже потом привлекать более сложные спектроскопические методы.
Многие органические соединения плавятся с разложением, что, как правило, легко обнаруживается по окрашиванию расплава или выделению газов. В этом случае температура разложения обычно характеризуется некоторым интервалом, сильно зависит от скорости нагревания (быстрое нагревание приводит к более высокому значению температуры разложения) и поэтому очень часто не может быть точно воспроизведена. Для некоторых веществ вообще нельзя говорить о точке фазового перехода, так как при сильном нагревании они обугливаются.
Между температурой плавления вещества и его молекулярным строением существует определенная зависимость. Замечено, что вещества с симметричными молекулами плавятся при более высокой температуре, чем вещества менее симметричного строения. Так, например, парафины нормального (неразветвленного) строения имеют более высокую температуру плавления, чем их изомеры с тем же числом атомов углерода. У стереоизомерных соединений транс-изомер, как правило, плавится при более высокой температуре [например, для малеиновой кислоты (цис-форма) т. пл. 130 °С, а для фумаровой кислоты (транс-форма) т. пл. 287 °С].
Температура плавления вещества растет с увеличением степени ассоциации молекул. Так, сложные эфиры, неспособные к образованию водородных связей, плавятся при значительно более низких температурах, чем соответствующие карбоновые кислоты.
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫПЛАВЛЕНИЯ В КАПИЛЛЯРЕ
Тонкоизмельченное и хорошо высушенное вещество слоем 2—4 мм помещают в запаянную с одного конца капиллярную трубку (диаметр ~1 мм). Для этого открытый конец капилляра погружают в пробу вещества и набранное в верхний конец капилляра вещество осторожно стряхивают на дно капилляра (или бросают капилляр заплавленным концом вниз несколько раз через длинную стеклянную трубку, вертикально установленную на жесткой подставке).
Температуру плавления веществ, склонных к возгонке, определяют в капиллярах, запаянных с обоих концов, запаянный капилляр с веществом надо полностью погружать в нагревательную баню.
Капилляр для определения температуры плавления в простейшем случае закрепляют на термометре (желательно, чтобы термометр был более точным) при помощи резинового колечка (рис. А. 110). Проба вещества должна находиться при этом на уровне ртутного шарика термометра. Затем в стакане с парафиновым или силиконовым маслом (теплопередающая среда, позволяющая проводить нагревание до -250 °С) медленно D—6 град/мин, а вблизи точки плавления — 1—2 град/мин) повышают температуру, доводя вещество до плавления.
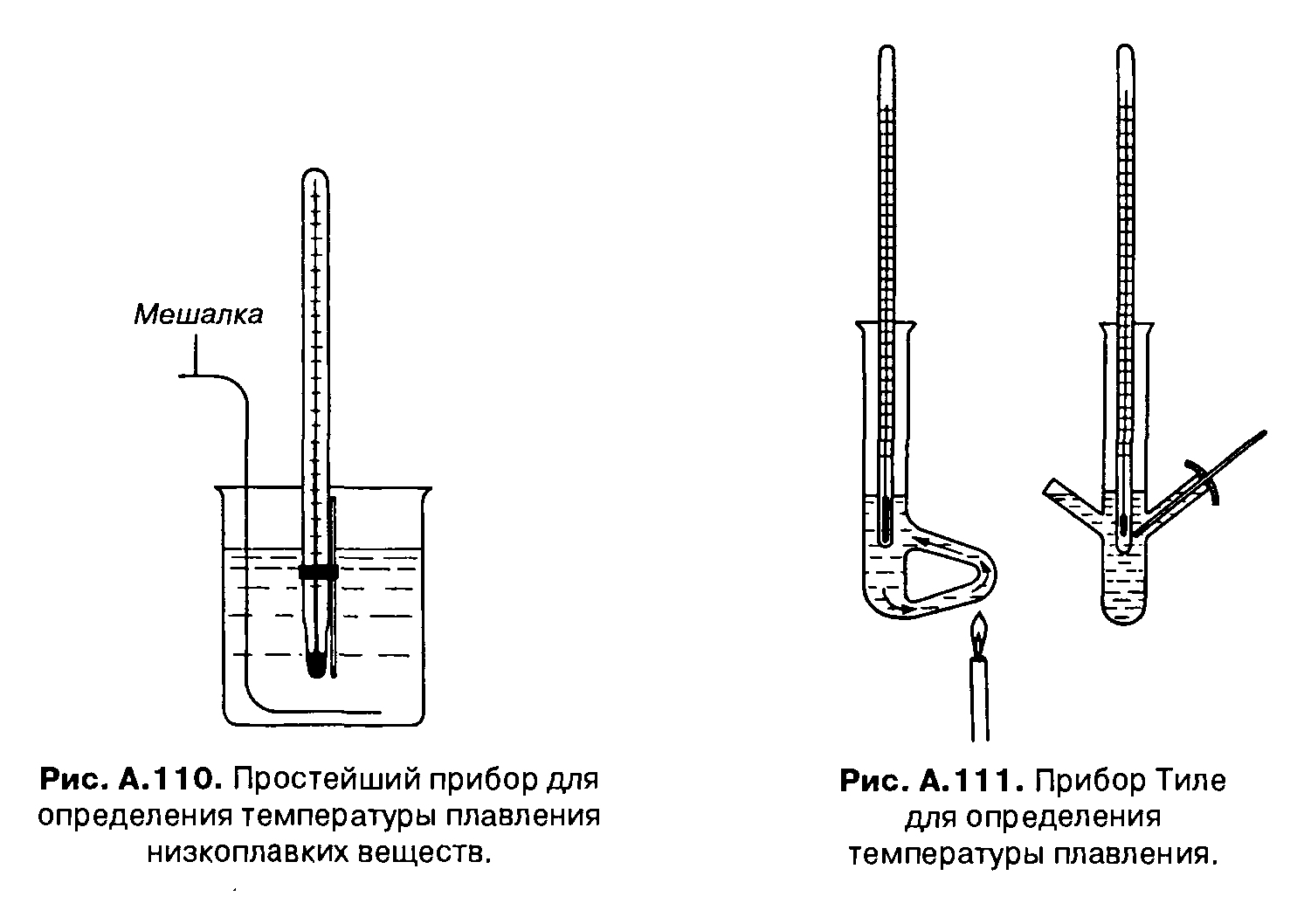
Удобнее определять температуру плавления в приборе Тиле (рис. А. 111), в котором более равномерен теплоперенос.
Температурой плавления при использовании описанной методики считается температура в момент полного расплавления вещества (просветвления расплава). Точность определения температуры плавления при этом составляет не более ±0,5 °С. Если испытывают не совсем чистое вещество, указывается температурный интервал от момента появления первых капель жидкой фазы до образования прозрачного расплава. Определенная так температура плавления (температурный интервал) обычно примерно на 1°С выше, чем установленная при помощи нагревательного столика.
Для определения температуры плавления высокоплавких соединений (т. пл. >250 °С) нагревание осуществляют в металлическом (медном или алюминиевом) блоке. Капилляр и термометр вводят в соответствующие отверстия блока, еще одно отверстие служит для наблюдения за процессом плавления. (Этоотверстие полезно закрыть слюдяными стеклами.)
Определение температуры плавления в капилляре возможно без особенно больших трудностей до -50 °С. В простейшем случае такие определения проводят в большом стеклянном стакане (рис. А110), наполненном охлаждающей смесью из сухого льда с метанолом. Сначала охлаждают вещество в капилляре до затвердевания, а затем при постоянном перемешивании дают охлаждающей смеси медленно нагреваться.
ОПРЕДЕЛЕНИЕ ТЕМПЕРАТУРЫПЛАВЛЕНИЯ С ПОМОЩЬЮ МИКРОСКОПА С НАГРЕВАТЕЛЬНЫМ СТОЛИКОМ
Использование оптического микроскопа с 50—100-кратным увеличением для изучения процесса плавления имеет ряд преимуществ по сравнению с определением температуры плавления в капилляре: количество вещества, необходимое для определения, крайне незначительно (достаточно нескольких миллиграммов или даже микрограммов). Все изменения, происходящие с веществом в процессе нагревания (отщепление воды от гидратов, полиморфные превращения, возгонку и разложение), можно отчетливо наблюдать под микроскопом (рис. А. 113).
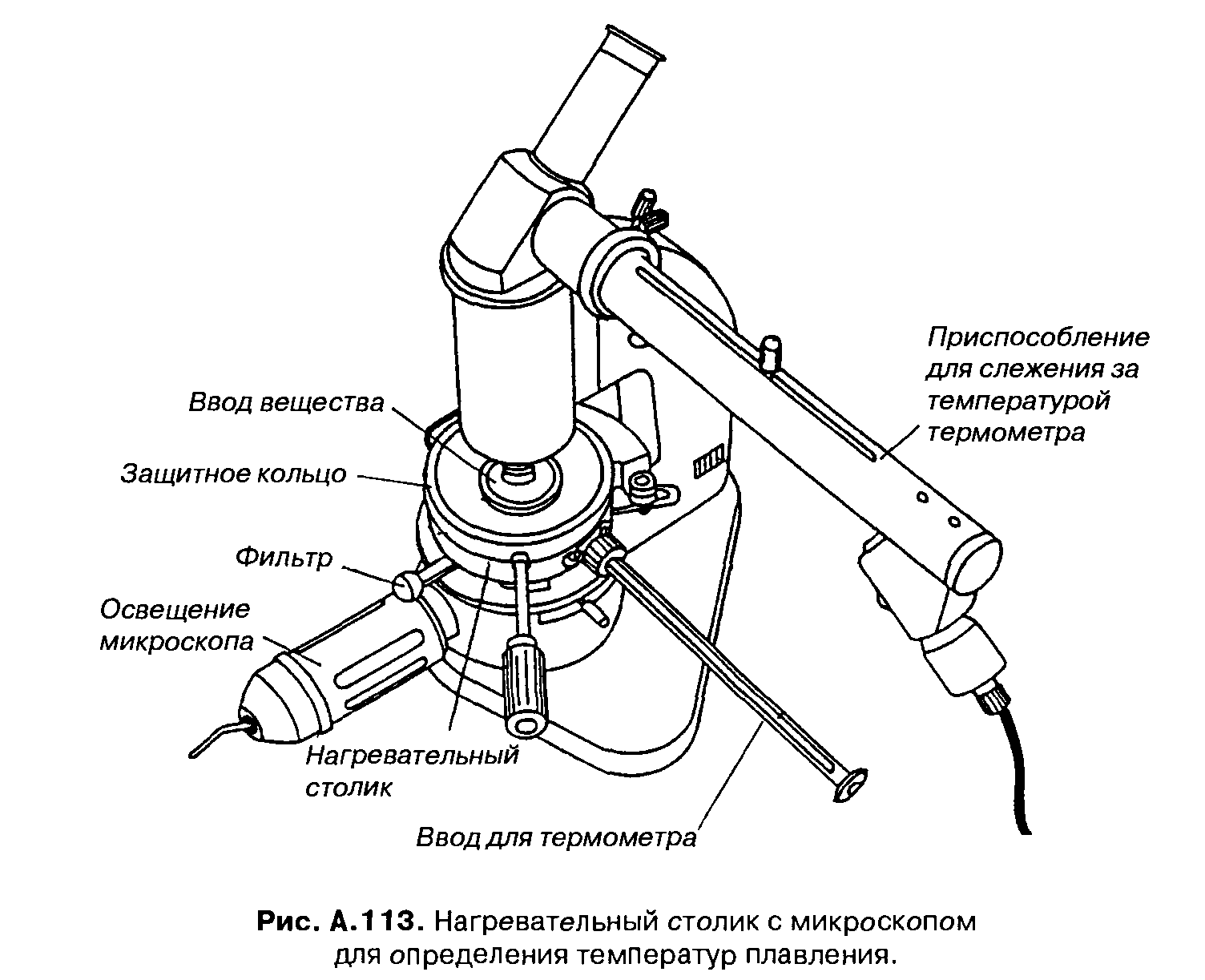
Дня этих целей микроскоп должен иметь специальный предметный столик (конструкции Кофлера и Бётиуса), в который вмонтирован электрический нагреватель; скорость нагревания последнего регулируется с помощью реостата. В боковое отверстие нагревательного столика помещают термометр, предварительно проградуированный на этом же столике с использованием подходящих соединений с известной температурой плавления; температуры плавления неизвестных веществ, полученные таким методом, уже содержат поправку на выступающий столбик.
Подготовка пробы к исследованию заключается во внесении нескольких кристалликов между специальными плоскими предметными и покровным стеклами. Большие кристаллы предварительно растирают между стеклами. Предметное стекло вместе с направляющим кольцом помещают на покрытый стеклянной пластиной нагревательный столик; изображение вещества должно находиться в поле зрения микроскопа.
Температуру плавления можно определять двумя различными способами. Согласно одному из них, температуру повышают непрерывно (вблизи точки плавления со скоростью 2—4 град/мин) до полного расплавления вещества. Началом плавления считают температуру, при которой начинают округляться углы и грани более крупных кристаллов. Точка, в которой кристаллы полностью исчезают, считается концом плавления вещества. Согласно второму способу, с помощью реостата устанавливают такую температуру, при которой достигается равновесие между твердой и жидкой фазами.
Температуру плавления сублимирующихся веществ определяют в плоских запаянных кюветах (кюветах Фишера).
При проведении пробы смешанного плавления на предметное стекло помещают по одному кристаллику каждого из двух испытуемых веществ и тщательно смешивают их, слегка нажимая и передвигая покровное стекло.
ТЕМПЕРАТУРА КРИСТАЛЛИЗАЦИИ (ЗАТВЕРДЕВАНИЯ)
Теоретически температуры плавления и затвердевания одинаковы, однако на практике это не так. Теоретическая температура— это температура, при которой жидкое вещество при охлаждении переходит в твердое (кристаллическое) состояние.
Для некоторых веществ идентичные или немного отличающиеся температуры плавления и затвердевания можно измерить, но для большинства органических соединений температура затвердевания оказывается ниже температуры плавления, причем разница иногда составляет 5-8°С. Это обусловлено переохлаждением. Переохлажденная жидкость (расплав) затвердевает очень быстро, что позволяет более четко определять температуры затвердевания по сравнению с температурой плавления. Это не означает, что температура затвердевания переохлажденной жидкости представляет собой характеристический параметр, поскольку степень переохлаждения зависит от внешних условий (в частности, от скорости охлаждения, формы кристалла и т. д.).
Температуру затвердевания определяют в простом приборе, показанном на рис. 15, если имеется достаточное количество вещества (4-5 г). Прибор состоит из двух пробирок, между которыми находится воздушная прослойка. Жидкость с термометром помещаются во внутреннюю пробирку. Для перемешивания используется проволока, скрученная в кольцо.
Для охлаждения можно использовать воду, смесь воды и льда, а также, если нужна температура ниже 0°С, смесь соли и льда или твердая углекислота. Во время охлаждения необходимо интенсивное перемешивание. Температурой затвердевания считают температуру, при которой жидкость, бывшая до этого светлой и прозрачной, становится мутной из-за появления мелких кристаллов.

Для определения температуры затвердевания можно также использовать капиллярную технику, но так как в этом случае перемешивание образца невозможно, капиллярным методом можно установить только температуру затвердевания переохлажденной жидкости.
Если в капилляр погрузить термопару, как описано выше для определения температуры плавления, то можно получить кривую охлаждения образца. Теплота кристаллизации вызовет появление излома на кривой.
Температуру замерзания можно также определить с помощью микроскопа с нагревательным столиком. Жидкость нагревают на несколько градусов выше температуры плавления, а затем медленно охлаждают. Температуру затвердевания определяют по появлению в жидкости первого кристалла. Часто случается, что первыми появляются не кристаллы исходной формы, а кристаллические агрегаты (сферолиты), когда иглы как бы растут из точки в расплаве. Часто образуются также дендритные структуры. Некоторые соединения не кристаллизуются при охлаждении, особенно если жидкость охлаждают быстро. Поскольку вязкость в этих условиях растет очень быстро и препятствует кристаллизации, жидкость при этом переходит в стеклообразное состояние.
Переохлаждение часто встречается у веществ, имеющих иглообразную форму кристаллов, и относительно редко у веществ, кристаллизующихся в форме пластинок.
Из-за переохлаждения температуру затвердевания в основном определяют для жиров, масел, воска и т. д., имеющих очень широкие интервалы температур плавления, которые часто невозможно точно определить. Температуры затвердевания, однако, можно наблюдать по помутнению жидкости. Точность таких измерений достаточна для технических целей.
Температуру затвердевания такого вещества измеряют либо с помощью прибора, изображенного на рис. 15, либо при записи кривой охлаждения с помощью термопары, помещенной в капилляр с образцом.
ТЕМПЕРАТУРА КИПЕНИЯ
Температура кипения вещества в отличие от температуры плавления очень сильно зависит от давления, и определить этот параметр не так просто. В большинстве случаев за температуру кипения вещества принимается температурный интервал при перегонке вещества. При этом погрешности определения связаны с перегревом паров и недостатками прибора (например, неправильное положение термометра, что искажает истинное значение температуры кипения. Кроме того, источниками погрешностей являются неправильное определение поправки на показания термометра или неправильное измерение давления (например, при ошибочных показаниях манометра на вакуумной установке). Поэтому в литературе часто для одного и того же вещества указываются разные температуры кипения.
Влияние загрязнений на температуру кипения сильно зависит от характера примесей. Так, значительное влияние оказывают остаточные количества легколетучего растворителя. Напротив, примесь вещества с той же самой температурой кипения вообще может не влиять (при идеальном поведении) на температуру кипения. Как правило, примеси в незначительных количествах оказываются для температуры кипения гораздо менее существенными, чем для температуры плавления.
По указанным причинам температура кипения является гораздо менее надежным способом идентификации и критерием чистоты вещества, чем температура плавления.
Размеры молекул и межмолекулярное взаимодействие — вот что в значительной мере обусловливает величину температуры кипения. Так, например, температура кипения нормальных парафинов (С4-С12) возрастает на 20-30 °С при увеличении длины углеродной цепи на один С-атом. Соединения с разветвленными молекулами, как правило, обладают более низкими температурами кипения, чем соответствующие соединения линейного строения.
При одинаковом числе атомов углерода температура кипения увеличивается в ряду, простые эфиры < альдегиды < спирты, так как межмолекулярное взаимодействие (ассоциация) возрастает в том же порядке (водородные связи в спиртах).
Температуру кипения можно точно определить с помощью эбуллиометров. Принцип их действия основан на том, что жидкость нагревается (в приборе с обратным холодильником) до кипения и измеряется температура. При соответствующей конструкции прибора исключаются тепловые потери и перегрев пара.
Однако для работы в таких приборах обычно необходимы большие количества вещества (не меньше нескольких миллилитров).
Если в распоряжении имеется достаточное количество жидкости (>10 мл), то проще всего снять в приборе для перегонки кривую кипения. При этом надо обратить внимание на то, чтобы шарик термометра полностью омывался парами, смачивался конденсирующейся жидкостью и не был погружен слишком глубоко в перегретый пар.
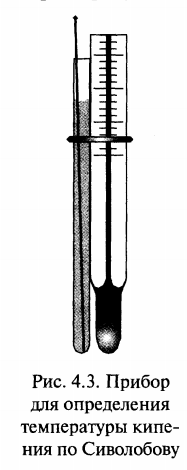
Температуру кипения малого количества вещества (0,2—1 мл) удобно определять по способу Сиволобова. На дно широкого тонкостенного заплавленного снизу капилляра диаметром 3—4 мм помещают каплю исследуемой жидкости. Затем в эту жидкость погружают другой, очень тонкий капилляр, заплавленный сверху (рис. 4.3). Капилляры прикрепляют к термометру и нагревают в приборе для определения температуры плавления (см. рис. 4.1). Вначале из внутреннего капилляра выделяются редкие пузырьки воздуха. Когда будет достигнута температура кипения, образуется цепочка пузырьков, поднимающаяся равномерной струей.
ОПРЕДЕЛЕНИЕ ПЛОТНОСТИ
Плотность вещества определяют, как правило, при 20 °С и относят ее к плотности дистиллированной воды при 4 °С. Поэтому в литературе обычно встречается обозначение плотности d420.Для измерения плотности применяют ареометры и пикнометры.
Плотность является постоянной величиной для каждого химически однородного вещества и для раствора определенной концентрации при данной температуре. Поэтому по величине плотности раствора можно находить его концентрацию, пользуясь специально составленными таблицами. Для большинства веществ плотность увеличивается с увеличением концентрации растворенного вещества, однако имеются случаи, когда с увеличением концентрации вещества плотность уменьшается. Таким редким исключением являются растворы аммиака.
Плотность зависит от температуры, при которой ее определяют. С повышением температуры она понижается, а с понижением температуры—повышается. Поэтому при определении плотности необходимо отмечать температуру, при которой производилось определение.
Ареометры представляют собой запаянные с обоих концов стеклянные трубки с расширением книзу, которое частично заполнено дробью (рис. 4.4, а, б). В верхней узкой части ареометра имеется шкала с делениями, позволяющая определять плотность с точностью до 0,001 г/см3. Испытуемую жидкость наливают в цилиндр, в который должен свободно погружаться ареометр, не касаясь стенок. Ареометр не следует выпускать из рук, пока не станет очевидно, что он плавает. Он не должен касаться дна. Отсчет производят по верхнему мениску жидкости. Удобны ареометры с вмонтированными в них термометрами, что позволяет одновременно с определением плотности фиксировать и температуру измерения.
Плотность вещества с точностью до 0,0001 г/см3 можно измерить с помощью пикнометра, который представляет собой маленькую мерную колбу вместимостью от 1 до 10 мл (рис. 4.4, в).
Чистый и сухой пикнометр взвешивают, заполняют дистиллированной водой, термостатируют при фиксированной температуре 15-30 минут. Потом воду доливают, чтоб уровень совпал с меткой на пикнометре, и вновь взвешивают. Воду удаляют и, заполнив высушенный пикнометр исследуемой жидкостью, определяют ее массу (также при фиксированной температуре). Отношение полученной массы к массе воды в объеме пикнометра, приведенной к 4 °С, соответствует плотности исследуемого вещества.
Плотность твердых порошкообразных тел определяют в волюмометрах. Волюмометрами называют пикнометры, применяемые для определения плотности порошкообразных твердых веществ. Такие пикнометры имеют емкость 50 мл. Если вещество, плотность которого надо определить, растворимо в воде, то выбирают жидкость, которая не растворяет исследуемое вещество. Жидкость должна хорошо смачивать исследуемое вещество. Перед определением плотности исследуемое вещество, измельченное до порошкообразного состояния, высушивают в сушильном шкафу в течение 1,5—2 час. при 105°, если эта температура допустима для взятого вещества.
Определение плотности начинают с определения плотности выбранной жидкости, способом, описанным выше. В тот же волюмометр, предварительно тщательно промытый, высушенный и взвешенный на аналитических весах, насыпают исследуемое порошкообразное вещество в количестве нескольких граммов, взвешивают и по разности весов точно определяют навеску взятого вещества.
Затем наливают в волюмометр небольшими порциями жидкость, каждый раз, тщательно перемешивая, содержимое. Когда прибор будет заполнен на 2/3, его помещают на 1—2 часа на водяную баню, нагретую до 60—65°, для удаления из порошкообразного вещества пузырьков воздуха. Время от времени содержимое волюмометра слегка взбалтывают. Когда выделение пузырьков будет закончено, прибор охлаждают, доливают до метки жидкостью и взвешивают. Таким образом определяют вес волюмометра с порошкообразным веществом и жидкостью. Для удаления пузырьков воздуха также можно использовать вакуум, но при этом надо следить, чтоб жидкость не кипела под вакуумом.
Плотность порошкообразного твердого тела (г/см3) определяют по формуле

где k — вес пикнометра, наполненного жидкостью, г;
р — вес порошкообразного вещества, г;
F — вес пикнометра с порошкообразным веществом и жидкостью, г;
γж—удельный вес жидкости, г/см3.
Правильные результаты по этому методу могут получиться только при условии, что из порошкообразного вещества будет удален весь воздух.
ОПРЕДЕЛЕНИЕ ПОКАЗАТЕЛЯ ПРЕЛОМЛЕНИЯ
Для идентификации жидких веществ и проверки их чистоты можно использовать также определение показателя преломления n. Если луч монохроматического света проходит через границу раздела двух сред (рис. А 114), то он отклоняется от первоначального направления по закону Снеллиуса
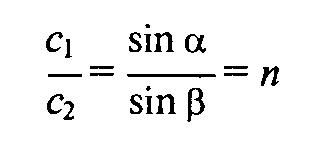
где c1 и с2 — скорости света в средах 1 и 2 В общем случае одной из сред служит воздух.

Показатель преломления сильно зависит от температуры. У органических жидкостей с ростом температуры на 1 °С он падает на 0,0005. Кроме того, показатель преломления сильно меняется в зависимости от длины волны света (дисперсия). Обычно указывается показатель преломления для спектральной линии желтого излучения натрия (D-линия, 589 нм). Температуру измерения и длину волны света (или спектральную линию) отмечают при значении показателя преломления соответственно верхним и нижним индексами, например nD25.
При определении показателя преломления газообразных веществ стандартной средой является вакуум, для жидких и твёрдых – воздух, так как различие между ним и вакуумом здесь становится несущественным.
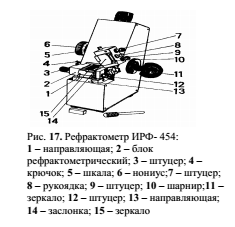
Определение показателя преломления осуществляют с помощью рефрактометров.
Одна из моделей рефрактометра (ИРФ-454) представлена на рис. 17.
ОПРЕДЕЛЕНИЕ УДЕЛЬНОГО ВРАЩЕНИЯ
Определенные химические соединения обладают «оптической активностью», т. е. если через них пропускать плоскополяризованный свет, то они поворачивают плоскость колебаний света на определенный угол — угол вращения α. Оптическая активность возникает в тех случаях, когда молекула соответствующего соединения хиральна.
Вращение плоскости поляризации может происходить вправо (обозначается знаком «+»; для наблюдателя — по часовой стрелке) или влево (обозначается знаком «-»). Для данного растворителя угол вращения α зависит от концентрации c (в граммах на 100 мл раствора), толщины слоя l вещества (в дециметрах), температуры t и длины волны λ'. Для данных длины волны и температуры угол вращения определяют по формуле
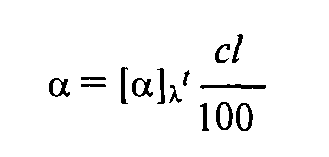
где [α]λ' — удельное вращение. Измерения обычно проводят для D-линии натрия при температурах 20 или 25 °С, что обязательно указывается, например [α]D20
Угол вращения α определяют с помощью поляриметров.
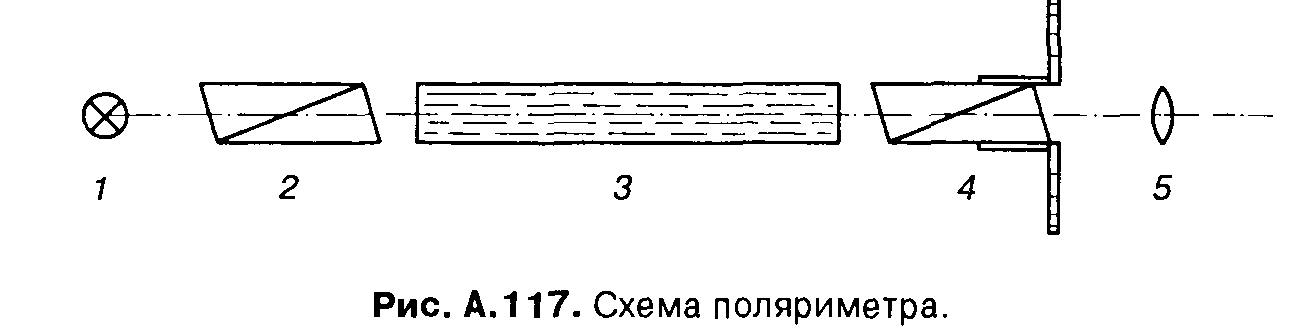
Принципиальная схема визуального поляриметра (рис. А. 117) включает источник монохроматического света 1, излучение которого поляризуется при прохождении через призму Николя 2 (поляризатор); далее поляризованный луч проходит через кювету 3 с раствором исследуемого вещества. Происходящее при этом отклонение плоскости поляризации света определяют с помощью второй, вращающейся призмы Николя 4 (анализатора), которая жестко связана с градуированной шкалой. Наблюдаемое при этом через окуляр 5 зрительное поле, разделенное на две или три части различной яркости, следует сделать равномерно освещенным. Величину необходимого для этого поворота анализатора 4 считывают со шкалы. Для проверки нулевой точки прибора проводят аналогичные измерения без исследуемого раствора.
Полученный таким образом угол вращения +α может в равной мере соответствовать как правому вращению на угол α (или 180°+α), так и левому вращению на угол 180°-α (или 360°-α). Точное направление вращения определяют поэтому при помощи повторного измерения, которое проводят либо с половинной толщиной слоя, либо с половинной концентрацией жидкости. Если при этом получают угол вращения α/2 (или α/2+90°), то можно считать, что вещество является правовращающим; если же новый угол вращения равен 90°-α/2 (или 180°-α/2), то имеется левовращающее вещество. Поскольку удельное вращение не очень сильно зависит от температуры, измерения обычно проводят, не термостатируя кювету. При более точных измерениях термостатирование необходимо.
Удельное вращение значительно зависит от растворителя, а в некоторых случаях и от концентрации вещества, так как растворенное вещество может взаимодействовать с растворителем. Поэтому, приводя данные по оптическому вращению, надо всегда указывать применяемый растворитель и концентрацию вещества в растворе, например [α]D25 27,3° в воде (с = 0,130 г/мл).
Поляриметрические измерения применяются не только для проверки чистоты оптически активных веществ, но и для определения количественного содержания их в растворах. Например, этим методом можно поляриметрически определять содержание сахара в растворах (сахариметр).
Хроматография
Хроматографические методы заключаются в разделении веществ между неподвижной (стационарной) фазой и подвижной (мобильной) фазой, протекающей через нее. Компоненты смеси веществ, нанесенные на стационарную фазу, удерживаются ею весьма различным образом и поэтому перемещаются с различной скоростью и могут быть таким образом разделены.
Подвижная фаза может быть жидкой или газообразной, поэтому методы называют жидкостная хроматография (ЖХ) или газовая хроматография (ГХ). Неподвижная фаза может быть твердым веществом или жидкостью, фиксированной на твердом носителе. Она находится в мелкозернистом виде в колонке (колоночная хроматография) или в тонком слое на инертном носителе или пластинке (тонкослойная хроматография). Специальная фильтровальная бумага может также служить в качестве неподвижной фазы (бумажная хроматография).
Предварительно использованы две физико-химические предпосылки для хроматографического разделения — различное распределение компонентов из-за их отличий в растворимости в обеих фазах и фракционная адсорбция из-за различной адсорбции компонентов на неподвижной фазе. В зависимости от преобладающего влияния говорят о распределительной хроматографии или адсорбционной хроматографии.
Оба процесса часто строго не разделимы, а действуют более или менее согласованно. Пористые твердые вещества, использованные как носители для жидких неподвижных фаз при распределительной хроматографии, могут обладать определенной адсорбционной способностью, и твердые адсорбенты для адсорбционной хроматографии, покрытые компонентами подвижной фазы или смесью веществ, могут способствовать их распределению.
Другие хроматографические методы разделения базируются на ионном обмене (на ионообменных смолах или гелях); при этом ионы могут быть разделены на пористых твердых веществах по величине молекулярной массы или величине биосродства (аффинная хроматография с неподвижными фазами, модифицированными определенными лигандами — ферментами, белками, липидами и др., способствующими селективной адсорбции). Информацию об этом можно найти в специальной литературе.
В качестве неподвижной фазы для адсорбционной хроматографии используются главным образом силикагель и оксид алюминия (нейтральный, кислый или основный), активность которых зависит от содержания воды. На этих полярных адсорбентах разделяемые вещества адсорбируются согласно их полярности. В качестве подвижных фаз используют углеводороды (пентан, гексан, гептан, изооктан), хлоруглеводороды (дихлорметан, хлороформ, тетрахлоруглерод), простые эфиры (тетрагидрофуран, диоксан) и ацетонитрил. Их элюирующая способность повышается соответственно их элюотропному ряду, табл. А.95. Состав мобильной фазы зависит от полярности разделяемых веществ. Сперва применяют неполярный растворитель, а затем повышают, если необходимо, его элюирующую способность путем добавления какого-либо более полярного растворителя.

Для распределительной хроматографии используют силикагель как носитель, на который наносят жидкость как стационарную фазу. Полярные и гидрофильные неподвижные фазы — это вода, этиленгликоль, этилендиамин, эфиры цианалканов или диметилсульфоксид. В качестве элюирующих веществ используют менее полярные вещества, не смешивающиеся с неподвижной фазой, прежде всего выше названные углеводороды или хлоруглеводороды. При этом, как и в хроматографии на полярных адсорбентах, быстрее всего элюируются наименее полярные вещества: алканы > галогенсодержащие соединения > простые эфиры > сложные эфиры > альдегиды > кетоны > спирты, амины > амиды > карбоновые кислоты.
Особенно проявили себя в этом отношении так называемые «обращенные фазы», это неполярные, гидрофобные неподвижные фазы (например, силикагель), поверхность которых модифицирована химически связанными алкильными остатками.
Такая модификация достигается обработкой поверхности силикагеля алкилхлорсиланами, например:
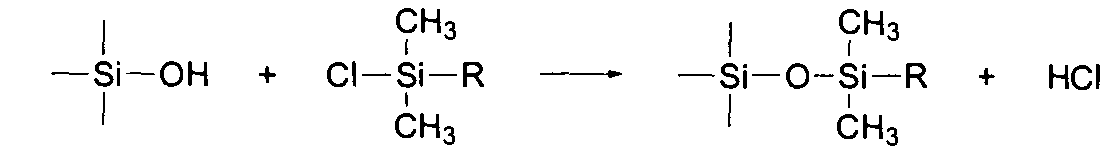
Чаще всего R — остаток октадецила С18Н37, но это могут быть и октил-, циклогексил- или фенильные остатки и замещенные алкилы с полярными циано-, нитро- или аминогруппами. Фазы, используемые для разделения энантиомеров, содержат хиральные группы, как, например, модифицированные целлюлозы, циклодекстрины (циклен из 6 или 8 глюкозных единиц) или другие асимметричные остатки. Хроматографическое разделение с «обращенными» фазами нельзя четко отнести к распределительной или адсорбционной хроматографии.
Обращенные фазы элюируют сильнополярными растворителями, как вода, или смесями вода—метанол или вода—ацетонитрил. При этом полярные вещества элюируются в обратном порядке по сравнению с хроматографией с полярными неподвижными фазами. Они элюируются тем быстрее, чем выше их полярность: карбоновые кислоты > спирты, фенолы > амины > сложные эфиры, альдегиды > кетоны > галогенсодержащие соединения.
При хроматографии смесей соединений, компоненты которых сильно отличаются по своей полярности, случается, что медленно идущие соединения выходят очень поздно или совсем не элюируются. Тогда принято в процессе хроматографии повысить элюирующую силу подвижной фазы непрерывным добавлением растворителя с повышенной элюирующей способностью (градиентное элюирование). В современных хроматографах это делается автоматически в заранее выбранных границах.
Разделяющая способность неподвижной фазы повышается при уменьшении величины частиц носителя, но при этом повышается сопротивление потоку. Для достижения достаточной скорости потока при величине частиц меньше 50 мкм подвижную фазу пропускают через неподвижную под давлением. Так как разделяющая способность при использовании мелкозернистых фаз мало зависит от скорости потока подвижной фазы, можно работать при высоких скоростях потока, а время разделения будет небольшим.
ТОНКОСЛОЙНАЯ ХРОМАТОГРАФИЯ
При тонкослойной хроматографии разделение веществ происходит на открытых колонках, неподвижная фаза которых состоит из тонкого слоя адсорбента, нанесенного на инертную подложку. В качестве слоя носителя служат плоские пластинки из стекла, алюминия или полиэфирных пленок. Как адсорбент используют силикагель или оксид алюминия, смешанные с гипсом в качестве связующего. Для получения однородных слоев одинаковой толщины применяют специальные приборы. Слои наносят в мокром состоянии, затем высушивают на воздухе и активируют при повышенной температуре 105—150 °С). Активность возрастает с понижением содержания воды. Активированные пластинки хранят в эксикаторе.
Удачно используют продажные приготовленные носители. Для восходящей хроматографии вырезают из нанесенной пластинки полоски нужной ширины (1,5 см на пробу) и около 10 см длиной. Для двумерной хроматографии берутся квадраты размером 10 х 10 см.
При хроматографировании смесей неизвестных веществ сначала используют циклогексан, затем толуол или хлороформ и в зависимости от полученных результатов переходят далее к полярным растворителям. Можно применять и смеси растворителей, например хорошие результаты дает смесь циклогексана с возрастающими добавками этилацетата 5, 10, 20% и более).
В предварительных опытах при подборе элюентов можно использовать радиальную хроматографию. При этом в центр пробы подают из микрокапилляра растворитель и оценивают его элюирующий эффект по образующимся концентрическим зонам.
Тонкослойная хроматография может быть восходящей, нисходящей, радиальной и двумерной. Наибольшее значение имеет удобная для выполнения восходящая ТСХ.
Вещества наносят в виде 1%-ного раствора в неполярном растворителе. Общее количество вещества целесообразно определять при помощи предварительного опыта. Слишком высокая концентрация разделяемых веществ приводит к появлению «хвостов» на хроматограмме и снижает эффективность разделения. Хорошо разделяемое количество вещества повышается с увеличением активности и толщины слоя адсорбента.
Растворы испытуемых смесей наносят на пластину с помощью тонкого капилляра (например, капилляра для определения температуры плавления); при погружении в раствор такие капилляры самопроизвольно заполняются под действием капиллярных сил. Нанесенные пятна испытуемых образцов должны быть расположены на расстоянии 1—2 см друг от друга, иметь небольшой размер (2-3 мм) и находиться не ближе 1,5 см от нижнего края пластины. Если раствор образца очень разбавленный, нанесение одного пятна повторяют несколько раз, дожидаясь после каждого нанесения испарения растворителя. Для двумерного хроматографирования пятно располагают в одном из углов квадратной пластины на расстоянии 1,5 см от краев.
Проявление хроматограммы проводят в плотно закрытой камере, атмосфера которой насыщена парами элюента (для более полного насыщения стенки камеры оклеивают полосками фильтровальной бумаги и оставляют стоять с элюентом в течение 30 мин). Пластину (полоску фольги) устанавливают в камере почти вертикально, погрузив конец в жидкость на ~5 мм.
Для предварительных опытов часто хроматограмму проявляют в прикрытом часовым стеклом химическом стакане, в который вложена манжета из фильтровальной бумаги.
В большинстве случаев для разделения достаточно подъема фронта элюента на высоту ~10 см. После этого пластину вынимают из камеры и сразу же с помощью острого предмета (шпателя, карандаша) отмечают положение фронта элюента.
Проявленные пластины высушивают на воздухе. Положение пятен некоторых бесцветных веществ, которые флуоресцируют, можно установить при рассмотрении пластин в УФ-свете. Если к адсорбенту добавлен флуоресцирующий индикатор, то все вещества, поглощающие в УФ-области, при освещении УФ-светом проявляются на флуоресцирующем поле в виде темных пятен.
В большинстве случаев удается проявить хроматографические пятна в присутствии паров иода. Для этого хроматограмму помещают в закрытый сосуд, в котором находится кристаллик иода. Вещество проявляется в виде коричневого пятна или (реже) при длительном действии паров иода в виде белого пятна на темном фоне. Для проявления хроматограммы применяют, кроме того, обработку парами брома, опрыскивание раствором перманганата калия и другими подходящими реагентами (табл. А.97) с применением распылителя (рис. А.98).

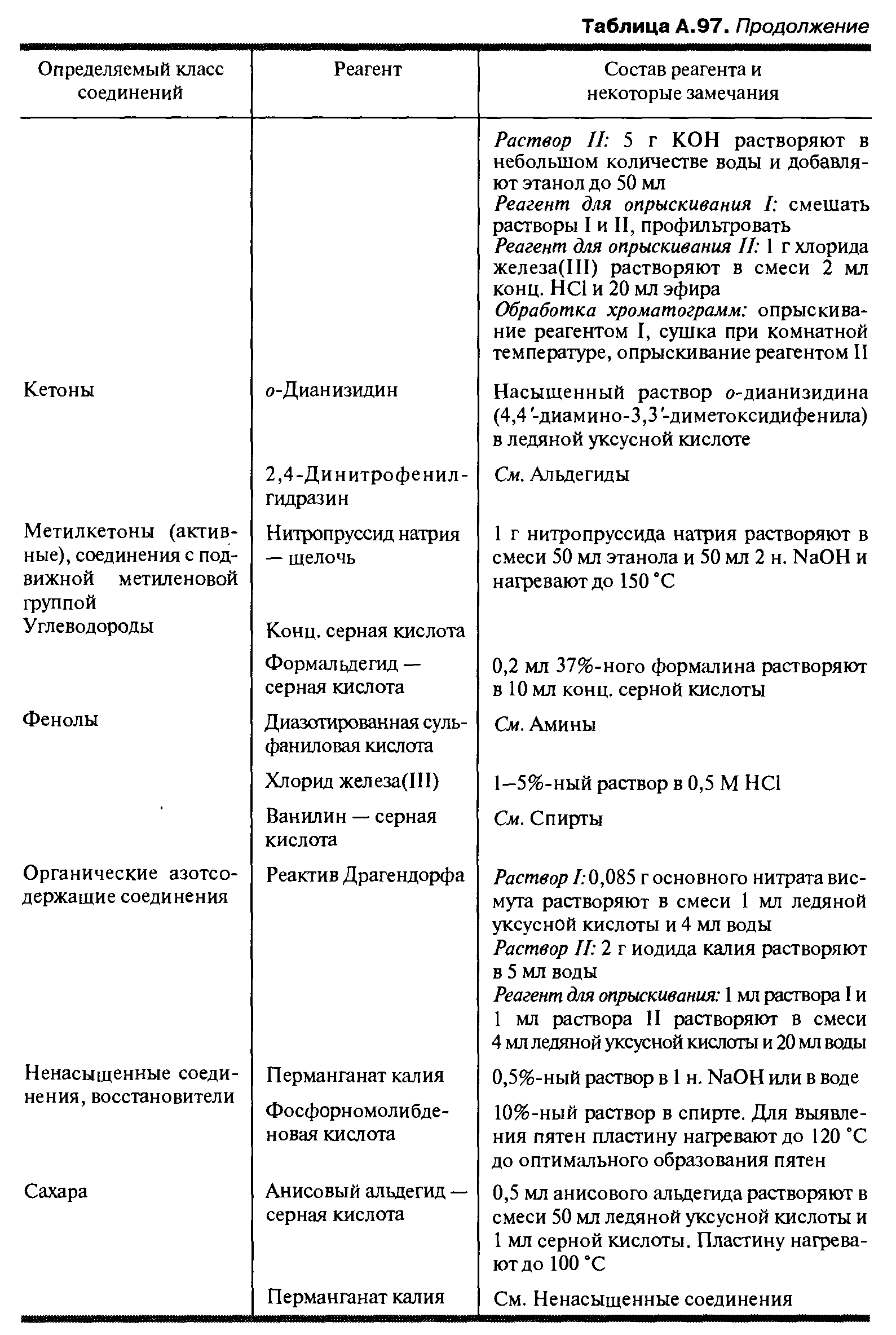

В общем случае