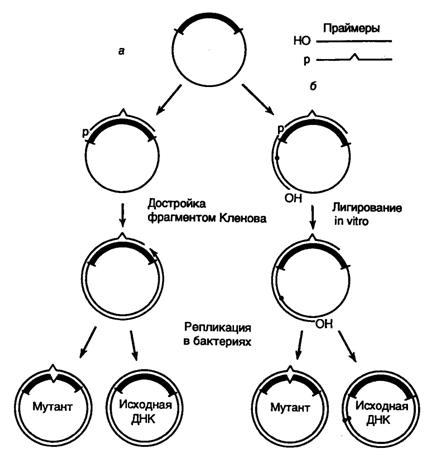
Рис. II.17. Направленный мутагенез с использованием олигонуклеотидов
а – система с одним мутагенизирующим праймером;
б – система с двумя праймерами и селектируемым маркером
В простейшем случае для проведения желаемой замены нуклеотидов синтезируют 15–20-членный олигодезоксирибонуклеотид, комплементарный одной из цепей ДНК исследуемого гена (рис. II.17, а). При этом в данный нуклеотид в процессе синтеза вносят требуемые замены нуклеотидов. Мутагенизируемый ген клонируют в фазмидном векторе, который переводят в одноцепочечную форму. После выделения такой одноцепочечной ДНК с клонированным геном дикого типа с ней гибридизуют синтетический олигонуклеотид и, используя его в качестве затравки, синтезируют с помощью фрагмента Кленова in vitro комплементарную цепь ДНК. Образующаяся в итоге двухцепочечная кольцевая молекула ДНК представляет собой гетеродуплекс, дочерняя цепь которого комплементарна исходной родительской цепи на всем протяжении, кроме участка, занимаемого олигонуклеотидом, где мутантный нуклеотид (нуклеотиды) некомплементарен соответствующему нуклеотиду матрицы.
Остающийся после завершения синтеза дочерней цепи ДНК одноцепочечный разрыв ликвидируют с помощью ДНК-лигазы, а образовавшуюся двухцепочечную ковалентно замкнутую ДНК вводят в бактериальные клетки, где она проходит несколько раундов репликации. После первого раунда репликации происходит сегрегация в разные молекулы ДНК мутантного аллеля и аллеля дикого типа исследуемого гена, и далее эти молекулы продолжают реплицироваться независимо друг от друга, что приводит к образованию двух популяций рекомбинантных ДНК, содержащих изучаемый ген в мутантной и исходной формах. Таким образом, теоретически каждая вторая молекула, выделенная из подобных бактериальных клеток, должна содержать в клонированном гене требуемую мутацию, локализованную в строго определенном сайте.
Для того чтобы отличить мутантные ДНК от нормальных, проводят либо прямое секвенирование мутагенизируемого участка гена, либо гибридизацию рекомбинантных молекул с олигонуклеотидом, ранее использованным в качестве затравки, предварительно меченным радиоактивными изотопами (например 32P). Связь этого олигонуклеотида, содержащего замены нуклеотидов, в гибриде будет более прочной с мутантной ДНК, чем с ДНК дикого типа, так как в первом случае олигонуклеотид и ДНК будут полностью комплементарны друг другу. Плавление гибрида и отделение олигонуклеотида от ДНК произойдут при более низких температурах, если гибрид был образован с ДНК дикого типа, а не с мутантной ДНК. Освободившиеся в результате плавления гибридов олигонуклеотиды отделяют от ДНК промыванием комплекса на фильтрах, что проявляется в отсутствие сигнала при авторадиографии, тогда как в случае гибридов олигонуклеотида и мутантной ДНК будет появляться четкий сигнал.
На практике, однако, частота возникновения мутантных ДНК в общей популяции рекомбинантных мутагенизируемых молекул бывает значительно > 50%. Это обусловлено рядом причин, основными из которых in vitro являются 3'®5'-экзонуклеазная активность фрагмента Кленова и его способность вытеснять цепь ДНК в направлении 5’®3’. Для преодоления первого затруднения мутантные нуклеотиды в олигонуклеотидном праймере располагают не ближе, чем за четыре нуклеотида от его 3’-конца (обычно за семь–десять нуклеотидов). Чтобы избежать последствий второго источника артефактов, для синтеза комплементарной цепи в мутагенизируемой одноцепочечной молекуле ДНК используют систему из двух праймеров (см. рис. II.17, б). В этой системе второй праймер выбирается таким образом, чтобы его 3’-конец после гибридизации с мутагенизируемой одноцепочечной молекулой ДНК располагался на расстоянии 1–3 т.п.о. от фосфорилированного 5’-конца мутагенизирующего нуклеотида. В этом случае после заполнения бреши между двумя олигонуклеотидами вновь синтезированной ДНК, синтез которой начался с дополнительного праймера, произойдет лигирование одноцепочечного разрыва ДНК-лигазой, которая присутствует в реакционной смеси вместе с фрагментом Кленова, поскольку 5’-конец мутагенизирующего нуклеотида фосфорилирован. Однако здесь не образуется кольцевой ковалентно замкнутой молекулы ДНК, так как на 5’-конце дополнительного праймера отсутствует фосфатная группа. После завершения реакции образовавшимися молекулами ДНК трансформируют клетки E. coli и производят отбор мутантных молекул ДНК в клонах бактерий по гибридизации с мутагенизирующим олигонуклеотидом, меченным радиоактивным изотопом.
Эффективность системы направленного мутагенеза с использованием двух олигонуклеотидов приближается к максимально теоретически возможной – 50%. С помощью этого метода можно получать все виды мутаций, как точковые с заменами одного–трех оснований, так и протяженные делеции длиной в 0,5–1,0 т.п.о., а также вставки, например для создания новых сайтов рестрикции. Если для получения точковых мутаций достаточно мутагенизирующего праймера длиной в 17–20 нуклеотидов, то в случае получения делеций праймеры должны быть длиннее (30–40 нуклеотидов), поскольку они удерживают последовательности матричной ДНК, фланкирующие выпетливаемый участок. Дальнейшее развитие системы направленного мутагенеза с использованием двух олигонуклеотидов, получившей название системы сопряженного праймирования, позволило значительно упростить процедуру отбора мутантных ДНК. В таких усовершенствованных системах в дополнительный олигонуклеотид вводится мутация, инактивирующая или восстанавливающая в дочерней мутантной ДНК какой-либо селектируемый маркер. В одной из таких систем, где в качестве вектора используется ДНК фага M13mp18amIV, селектируемым маркером служит амбер-мутация в гене IV. Матричную ДНК получали, выращивая мутантные фаги M13mp18amIV в бактериальных клетках E. coli TG1, содержащих ген-супрессор амбер-мутаций Su2+. При этом данный фаг не развивается в клетках несупрессорных штаммов E. coli Su2-.
Для проведения мутагенеза одноцепочечную ДНК фага M13mp18am1Y гибридизуют с двумя праймерами, один из которых мутагенизирующий, а другой, комплементарный гену IV, содержит точковую мутацию в амбер-кодоне, восстанавливающую последовательность нуклеотидов дикого типа. После достройки комплементарной цепи ДНК фага M13mp18am1Y с использованием вышеупомянутых праймеров двухцепочечные ДНК вводят в бактериальные клетки Su2-, которые, кроме того, дефектны по системе репарации. Поскольку бактериальные клетки, дефектные по системе репарации, сами по себе обладают мутаторным фенотипом (т.е. для них характерна повышенная частота возникновения спонтанных мутаций), то для того чтобы в мутируемые ДНК не вносить дополнительные мутации, трансформированные бактериальные клетки высевают на газон клеток E. coli Su2- с нормальной системой репарации. При таком подходе фаговые частицы проходят лишь первый цикл развития в клетках с двумя мутациями, а все последующие – в клетках Su2- с одной мутацией. В результате в таких клетках образуются только те фаговые частицы, ДНК которых содержат сайт-специфическую мутацию и восстановленный ген IV.
Аналогичный подход был использован в системе с геном b-галактозидазы, находящимся в составе векторов серии M13mp. В такой системе фаговый ген кодирует a-пептид b-галактозидазы E. coli и комплементирует (восстанавливает) дефектную b-галактозидазу бактериальных клеток-хозяев. Введение амбер-мутации в фаговый ген b-галактозидазы с помощью праймера, используемого в паре с олигонуклеотидом-мутатором, сопровождается потерей дочерними фаговыми частицами способности комплементировать мутантную b-галактозидазу бактериальных клеток. В результате в образующихся фаговых бляшках отсутствует активность b-галактозидазы, которая бы расщепляла искусственный субстрат BCIG с образованием продукта, окрашенного в голубой цвет. Вследствие этого бляшки, образованные фаговыми частицами, содержащими ДНК с сайт-специфическими мутациями, не окрашены и их легко отбирать визуально.
Хорошо себя зарекомендовали и системы циклического отбора мутантных ДНК, содержащие сайт-специфические мутации, в которых используются свойства систем рестрикции–модификации EcoK и EcoB E. coli. Используемые для трансформации штаммы E. coliмогут содержать системы рестрикции–модификации EcoK- или EcoB-типа. В том случае, если родительскую цепь ДНК для мутагенеза выделяют из клеток, дефектных по метилазам системы модификации, то после достройки комплементарной цепи с мутантного олигонуклеотида такая неметилированная ДНК будет расщепляться системой рестрикции после введения в соответствующий бактериальный штамм. Если одновременно с олигонуклеотидом-мутатором использовать второй олигонуклеотид, изменяющий сайт рестрикции, то лишь ДНК с мутировавшим сайтом рестрикции в результате ошибочного функционирования системы репарации сохранится в бактериальных клетках. Такие ДНК с высокой вероятностью будут содержать и требуемую мутацию.
Недавно была описана система, в которой в родительскую цепь ДНК фазмиды вводили вместо остатков тимидина остатки уридина. В этом случае после синтеза дочерней комплементарной ДНК in vitro с использованием мутантного олигонуклеотида в качестве затравки родительская цепь ДНК инактивируется в трансформированных бактериальных клетках с помощью эндогенной урацил-N-гликозилазы, удаляющей из ДНК остатки урацила, что сопровождается частичным гидролизом этой цепи с последующей репарацией брешей по мутантной цепи в качестве матрицы и закреплением введенной мутации.
Наконец, следует упомянуть систему олигонуклеотид-зависимого сайт-специфического мутагенеза, в которой для отбора мутантных цепей ДНК используется попеременно то один, то другой селектируемый маркер, в качестве которых выступают гены устойчивости к антибиотикам. Это позволяет вводить с высокой эффективностью в один и тот же ген несколько сайт-специфических мутаций в последовательных циклах направленного мутагенеза. В таких системах в первом цикле вместе с мутагенизирующим праймером в качестве селектируемого используют праймер, вносящий мутацию, которая придает бактериальным клеткам устойчивость к одному антибиотику, а во втором цикле – к другому (с восстановлением чувствительности к первому антибиотику). Далее такие циклы могут быть повторены необходимое число раз, которое определяется количеством требуемых мутаций.
Разработка систем сопряженного праймирования позволила значительно увеличить эффективность направленного мутагенеза и довести выход ДНК, содержащих сайт-специфические мутации, до 70% от общего числа образующихся молекул ДНК.
Рассмотренные подходы к получению сайт-специфических мутаций с помощью олигонуклеотидов позволяют с высокой точностью и эффективностью производить замены отдельных нуклеотидов в строго определенных локусах. Однако чаще всего невозможно предсказать фенотипические последствия мутационных замен отдельных аминокислот в полипептидных цепях белков, и для получения необходимых фенотипических изменений требуется введение множественных мутаций в определенные участки гена с последующим отбором мутантов требуемого фенотипа. Для решения подобных задач был разработан и эффективно используется метод кассетного мутагенеза.
При реализации такого подхода из гена, клонированного в составе векторной плазмиды, по двум близко расположенным уникальным сайтам рестрикции вырезается фрагмент ДНК, в который необходимо внести мутации, и на его место встраивается синтетический двухцепочечный олигонуклеотид, содержащий необходимые замены нуклеотидов (кассету мутаций). В этом случае, если в окрестностях мутагенизируемого локуса гена отсутствуют подходящие природные сайты рестрикции, их вводят с помощью направленного мутагенеза. Разработка автоматических синтезаторов ДНК сделала синтез олигодезоксирибонуклеотидов простой и даже рутинной процедурой. Более того, использование на определенных этапах синтеза вместо одного нуклеотида смеси из двух, трех или даже всех четырех дезоксирибонуклеозидтрифосфатов позволяет получать за один прием сложную смесь олигонуклеотидов, которые могут содержать в определенных сайтах наборы кодонов для многих или даже для всех 20 природных аминокислот. Это дает возможность осуществлять одновременный скрининг по искомому мутантному фенотипу большого числа разных мутантных клонов, полученных в одном цикле клонирования. С помощью кассетного мутагенеза можно легко исследовать функциональную роль отдельных сайтов и целых доменов в полипептидных цепях конкретных белков и создавать рекомбинантные белки с новыми, подчас неожиданными свойствами.
Внесение множественных мутаций в разные участки полипептидных цепей исследуемых белков позволяет при наличии соответствующих эффективных методов отбора получать белки с требуемыми свойствами и уже после этого определять, какие именно аминокислоты придают такие свойства белку. Альтернативным методом исследования функциональной значимости отдельных аминокислот в белках является их целенаправленная замена на нейтральную аминокислоту, например аланин. Такая последовательная замена аминокислот в полипептидных цепях изучаемых белков на остатки аланина получила название сканирование аланином. Введение аланина в полипептидные цепи не изменяет их общей конформации, как это имеет место, например в случае замен на глицин или пролин, и не сопровождается ярко выраженными электростатическими или стерическими эффектами. Кроме того, аланин часто встречается в полипептидных цепях и с одинаковой частотой представлен как на внутренних, так и на внешних участках полипептидных цепей белковых глобул. С помощью сканирования аланином можно локализовать аминокислоты, образующие активный центр ферментов, исследовать участки полипептидных цепей, существенные для взаимодействия белков с другими макромолекулами и низкомолекулярными лигандами, изучать структуру эпитопов полипептидных цепей, а также ряд других структурных и функциональных особенностей белков.
Еще одним универсальным методом замены определенных аминокислот в полипептидных цепях белков in vivo является использование аминоацилированных различными аминокислотами супрессорных тРНК, которые узнают нонсенс-триплеты в мутантных мРНК в процессе трансляции. В результате в соответствующее место полипептидной цепи взамен аминокислоты, присутствующей в белке дикого типа, встраивается аминокислота, которую несет аминоацилированная супрессорная тРНК. В настоящее время в дополнение к природным супрессорным тРНК E. coli синтезированы in vitro гены, кодирующие супрессорные РНК новой специфичности. В итоге до 13 аминокислотных замен может быть произведено в синтезирующейся in vivo полипептидной цепи в результате супрессии кодона UAG (amber). С использованием такого подхода, в частности, удалось произвести >1600 замен аминокислот Lac-репрессора E. coli и локализовать участки полипептидной цепи, существенные для связывания индуктора, прочного связывания оператора и термостабильности белка.
Важным преимуществом метода амбер-супрессии перед другими методами направленного мутагенеза у бактерий и бактериофагов является то, что он не требует синтеза большого числа мутантных генов и их последующего отбора, так как введение одного мутантного гена в составе экспрессирующего вектора в клетки разных супрессорных штаммов E. coli позволяет получать разные замены аминокислот с одновременной сверхпродукцией мутантного белка в бактериальных клетках. Это облегчает его последующую очистку и изучение биохимических свойств.