· ЭКГ при выполнении которой можно выявить перегрузку различных отделов сердца, изменения миокарда
· обзорная рентгенография органов грудной клетки, на которой можно отметить изменение конфигурации сердца
· ЭХО-КГ - основная методика - позволяет увидеть морфологию порока и определить функциональное состояние сердца.
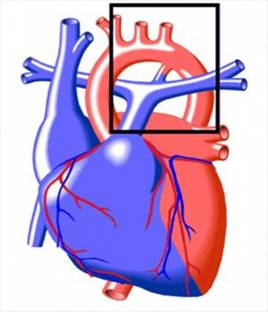 Открытый артериальный проток(ОАП) – наличие сообщения между аортой и лёгочной артерией, считающегося аномалией в постнатальном периоде (рис.1). Анатомическое закрытие или облитерация происходит в течение первых 2-х недель постнатальной жизни. Такие патологические состояния периода новорожденности как синдром дыхательных расстройств, врождённая пневмония, асфиксия в родах препятствуют закрытию ОАП. Объём крови, сбрасываемый из аорты в лёгочную артерию, приводит к развитию диастолической перегрузки и дилатации левых отделов сердца, особенно левого предсердия, гиперволемии в лёгких с формированием легочной гипертензии.
Открытый артериальный проток(ОАП) – наличие сообщения между аортой и лёгочной артерией, считающегося аномалией в постнатальном периоде (рис.1). Анатомическое закрытие или облитерация происходит в течение первых 2-х недель постнатальной жизни. Такие патологические состояния периода новорожденности как синдром дыхательных расстройств, врождённая пневмония, асфиксия в родах препятствуют закрытию ОАП. Объём крови, сбрасываемый из аорты в лёгочную артерию, приводит к развитию диастолической перегрузки и дилатации левых отделов сердца, особенно левого предсердия, гиперволемии в лёгких с формированием легочной гипертензии.
 Клиническая картина при ОАП является характерной для ВПС, протекающих с обогащением малого круга кровообращения, и будет зависеть от размера протока.
Клиническая картина при ОАП является характерной для ВПС, протекающих с обогащением малого круга кровообращения, и будет зависеть от размера протока.
 При естественном течении продолжительность жизни больных составляет 20-25 лет. После 12-месячного возраста редко происходит спонтанное закрытие артериального протока. Основными осложнениями ОАП являются сердечная недостаточность, лёгочная гипертензия, инфекционный эндокардит, аневризма аорты и\или лёгочной артерии.
При естественном течении продолжительность жизни больных составляет 20-25 лет. После 12-месячного возраста редко происходит спонтанное закрытие артериального протока. Основными осложнениями ОАП являются сердечная недостаточность, лёгочная гипертензия, инфекционный эндокардит, аневризма аорты и\или лёгочной артерии.
 Оперативное лечение подразумевает перевязку или пересечение с ушиванием аортального и лёгочного концов протока, но в последнее время применяется и катетерная эндоваскулярная окклюзия протока (рис.2).
Оперативное лечение подразумевает перевязку или пересечение с ушиванием аортального и лёгочного концов протока, но в последнее время применяется и катетерная эндоваскулярная окклюзия протока (рис.2).
|
|
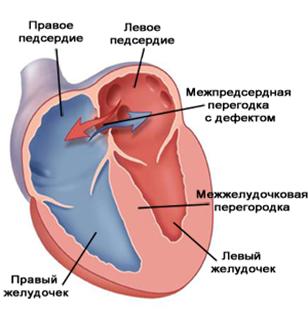 Дефект межпредсердной перегородки(ДМПП) - это группа пороков сердца, для которых характерно аномальное сообщение между предсердиями (рис.3).
Дефект межпредсердной перегородки(ДМПП) - это группа пороков сердца, для которых характерно аномальное сообщение между предсердиями (рис.3).
 Гемодинамические изменения при ДМПП развиваются постепенно вследствие разницы в давлениях в большом и малом кругах кровообращения, из-за чего формируется лево-правый сброс крови через дефект. Из-за поступления избыточного количества крови в правое предсердие и правый желудочек со временем развиваются их дилятация, гипертрофия. Формируется гемодинамический (относительный) стеноз лёгочной артерии, шум которого выслушиваться при аускультации. Изменения в сосудах малого круга кровообращения происходят по общим законам, но гораздо более медленно, чем при ДМЖП.
Гемодинамические изменения при ДМПП развиваются постепенно вследствие разницы в давлениях в большом и малом кругах кровообращения, из-за чего формируется лево-правый сброс крови через дефект. Из-за поступления избыточного количества крови в правое предсердие и правый желудочек со временем развиваются их дилятация, гипертрофия. Формируется гемодинамический (относительный) стеноз лёгочной артерии, шум которого выслушиваться при аускультации. Изменения в сосудах малого круга кровообращения происходят по общим законам, но гораздо более медленно, чем при ДМЖП.
 Заподозрить ДМПП в периоде новорожденности сложно. Признаки недостаточности кровообращения развиваются, как правило, значительно позже – на 1-3 годах жизни, когда происходит увеличение двигательной активности ребенка. Показатели физического развития у детей с ДМПП, как правило, соответствуют возрастной норме. Для пациентов раннего возраста характерны частые респираторные заболевания, сопровождающиеся бронхообструкцией. Признаки высокой лёгочной гипертензии развиваются поздно – к 16-25 годам.
Заподозрить ДМПП в периоде новорожденности сложно. Признаки недостаточности кровообращения развиваются, как правило, значительно позже – на 1-3 годах жизни, когда происходит увеличение двигательной активности ребенка. Показатели физического развития у детей с ДМПП, как правило, соответствуют возрастной норме. Для пациентов раннего возраста характерны частые респираторные заболевания, сопровождающиеся бронхообструкцией. Признаки высокой лёгочной гипертензии развиваются поздно – к 16-25 годам.
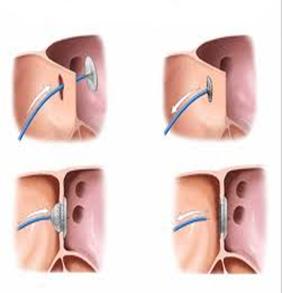
 Лечение сердечной недостаточности проводится по общим принципам. Хирургическое лечение заключается в радикальной коррекции – пластике ДМПП. Возможно эндоваскулярное закрытие ДМПП (рис.4).
Лечение сердечной недостаточности проводится по общим принципам. Хирургическое лечение заключается в радикальной коррекции – пластике ДМПП. Возможно эндоваскулярное закрытие ДМПП (рис.4).
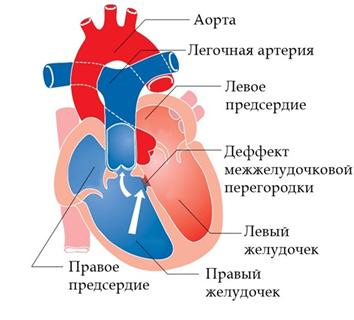 Дефект межжелудочковой перегородки(ДМЖП) - встречается наиболее часто, причём как в изолированном виде, так и в составе многих других пороков сердца (рис.5).
Дефект межжелудочковой перегородки(ДМЖП) - встречается наиболее часто, причём как в изолированном виде, так и в составе многих других пороков сердца (рис.5).
|
|
 В межжелудочковой перегородке выделяют 3 отдела: верхняя часть – мембранозная, прилегает к центральному фиброзному телу, средняя часть – мышечная, и нижняя – трабекулярная. Соответственно этим отделам называют и дефекты межжелудочковой перегородки, однако большинство из них имеют перимембранозную локализацию.
В межжелудочковой перегородке выделяют 3 отдела: верхняя часть – мембранозная, прилегает к центральному фиброзному телу, средняя часть – мышечная, и нижняя – трабекулярная. Соответственно этим отделам называют и дефекты межжелудочковой перегородки, однако большинство из них имеют перимембранозную локализацию.
Для правильной оценки величины дефекта его размер надо сравнивать с диаметром аорты. Мелкие дефекты размером 1-2 мм, расположенные в мышечной части МЖП, называются болезнью Толочинова–Роже. Внутрисердечные гемодинамические нарушения при ДМЖП начинают формироваться спустя некоторое время после рождения, как правило, на 3-5 сутки жизни. В раннем неонатальном периоде шум в сердце может отсутствовать вследствие одинакового давления в правом и левом желудочках из-за так называемой неонатальной легочной гипертензии. Постепенное падение давления в системе легочной артерии и в правом желудочке создаёт разность (градиент) давлений между желудочками, вследствие чего появляется сброс крови слева-направо (из области высокого давления в область низкого давления). Дополнительный объём крови, поступающий в правый желудочек и легочную артерию, приводит к переполнению сосудов малого круга кровообращения, где развивается легочная гипертензия.
Если хирургическая коррекция ВПС не проводится, начинают формироваться процессы склерозирования сосудов легких (высокая легочная гипертензия – синдром Эйзенменгера). Этот патологический процесс не имеет обратного развития и приводит к значительному повышению давления в легочной артерии (иногда до 100-120 мм.рт.ст.). В клинической картине заболевания отмечается множество патологических признаков: сердечный «горб», расширение границ относительной сердечной тупости, больше вправо. Самым характерным признаком синдрома Эйзенменгера является постепенное нарастание цианоза, - сначала периферического, а в дальнейшем и диффузного. Это происходит вследствие перекрестного сброса крови в области дефекта межжелудочковой перегородки, который при превышении давления в правом желудочке становится право-левым, т.е. меняет свое направление.
|
|
Клиническая картина при ДМЖП заключается в симптомокомплексе сердечной недостаточности, развивающейся на 1-3 месяцах жизни (в зависимости от размеров дефекта). Кроме признаков сердечной недостаточности ДМЖП может манифестировать пневмониями.
Оперативные вмешательства подразделяются на паллиативные операции (сужение лёгочной артерии) и радикальную коррекцию порока – пластику дефекта межжелудочковой перегородки.
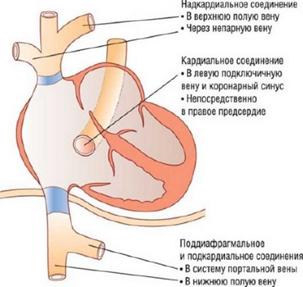
Аномальный дренаж легочных вен(АДЛВ) - врождённый порок сердца, для которого характерно впадение лёгочных вен в правое предсердие или в полые вены большого круга кровообращения. В норме все четыре лёгочные вены дренируются в левое предсердие. При частичном аномальном дренаже лёгочных вен одна или две лёгочные вены впадают в правое предсердие (как варианты – в верхнюю полую вену, нижнюю печеночную или воротную вену). При тотальном аномальном дренаже все лёгочные вены дренируются неправильно (рис.6). При этом возникает разной степени выраженности застой в малом круге кровообращения и перегрузка правого предсердия, которое значительно увеличивается. Для разгрузки последнего обязательным условием является наличие дефекта межпредсердной перегородки, через который кровь из правого предсердия будет поступать в левое предсердие, неся туда смешанную кровь. В классическом варианте встречается большой вторичный дефект межпредсердной перегородки.
Клиническая картина зависит от разновидности порока и, в первую очередь, от количества аномально дренирующихся вен. При тотальном АДЛВ клиническая картина специфична и развивается вскоре после рождения: одышка прогрессирует и становится резко выраженной, имеет место слабо выраженный цианоз, быстро развиваются и нарастают симптомы бивентрикулярной сердечной недостаточности, отмечается склонность к развитию тяжёлых пневмоний на фоне выраженной гиперволемии малого круга кровообращения. Дети, как правило, значительно отстают в физическом развитии.
 При частичном АДЛВ клиническая картина развивается медленнее и порок может обнаруживаться случайно, чаще всего на втором году жизни. Дети, как правило, развиваются удовлетворительно. Может отмечаться склонность к бронхо-лёгочным заболеваниям.
При частичном АДЛВ клиническая картина развивается медленнее и порок может обнаруживаться случайно, чаще всего на втором году жизни. Дети, как правило, развиваются удовлетворительно. Может отмечаться склонность к бронхо-лёгочным заболеваниям.
 При тотальном аномальном дренаже лёгочных вен больные погибают от рефрактерной сердечной недостаточности и высокой лёгочной гипертензии. Средняя продолжительность жизни – 2-6 месяцев. Частичный аномальный дренаж лёгочных вен характеризуется более спокойным прогнозом.
При тотальном аномальном дренаже лёгочных вен больные погибают от рефрактерной сердечной недостаточности и высокой лёгочной гипертензии. Средняя продолжительность жизни – 2-6 месяцев. Частичный аномальный дренаж лёгочных вен характеризуется более спокойным прогнозом.
Хирургическая коррекция проводится в различные возрастные периоды и зависит от типа ВПС. Операционная летальность при этом пороке составляет до 25% в периоде новорожденности и прогрессивно уменьшается с возрастом ребенка. Новорожденным иногда проводят паллиативную операцию - баллонную атриосептотомию с целью расширения межпредсердного сообщения. Радикальная операция заключается в создании широкого анастомоза лёгочных вен с левым предсердием, закрытии ДМПП.
Тетрада Фалло(ТФ) относится к наиболее распространённым порокам сердца синего типа (рис.7). При классическом варианте тетрады Фалло обнаруживается 4 признака: сужение выводного отдела правого желудочка на различных уровнях, дефект межжелудочковой перегородки, который всегда является большим, высоким, перимембранозным, гипертрофия миокарда правого желудочка и декстрапозиция аорты. Порок относится к ВПС цианотического типа с обеднением малого круга кровообращения.
Выделяют три клинико-анатомических варианта порока: 1) ТФ с атрезией устья лёгочной артерии – «крайняя», цианотическая форма; 2) классическая форма; 3) ТФ с минимальным стенозом легочной артерии, или «бледная», ацианотическая форма ТФ. Выделяют триаду Фалло, когда отсутствует дефект межжелудочковой перегородки. Тетрада Фалло может сочетается и с другими ВПС: при одновременном присутствии ДМПП, вариант называется пентадой Фалло. Наиболее часто ТФ сочетается с ОАП, за счёт которого происходит компенсаторное кровоснабжение лёгких. При «крайней» форме ТФ порок является «дуктус»-зависимым.

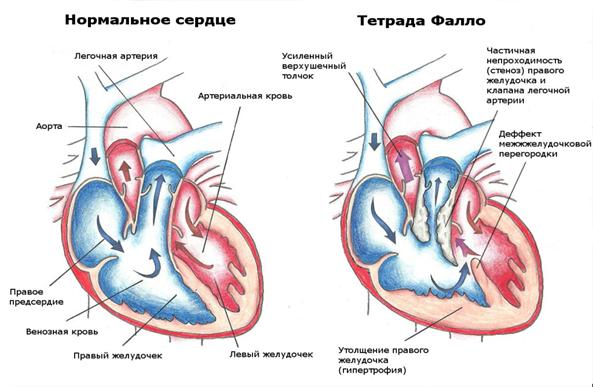 Степень нарушения гемодинамики определяется выраженностью стеноза легочной артерии. Обструкция может находиться на уровне выходного отдела правого желудочка, на уровне клапана лёгочной артерии, по ходу ствола и ветвей легочной артерии и на нескольких уровнях одновременно. Во время систолы кровь поступает из обоих желудочков в аорту и в меньшем количестве – в лёгочную артерию. Вследствие того, что аорта широкая и смещена вправо, кровь по ней проходит беспрепятственно, поэтому при классической форме тетрады Фалло никогда не бывает недостаточности кровообращения. Из-за большого дефекта межжелудочковой перегородки давление в обоих желудочках одинаковое. Степень гипоксии и тяжесть состояния больных коррелируют со степенью стеноза лёгочной артерии. У пациентов с крайней формой тетрады Фалло кровь в лёгкие поступает через открытый артериальный проток либо через коллатерали (артериовенозные анастомозы и бронхиальные артерии), которые могут развиваться внутриутробно, но чаще - постнатально. Компенсация кровообращения происходит за счет: 1) гипертрофии правого желудочка; 2) функционирования ОАП и/или коллатералей; 3) развития полиглобулии и полицитемии в результате длительной гипоксии;
Степень нарушения гемодинамики определяется выраженностью стеноза легочной артерии. Обструкция может находиться на уровне выходного отдела правого желудочка, на уровне клапана лёгочной артерии, по ходу ствола и ветвей легочной артерии и на нескольких уровнях одновременно. Во время систолы кровь поступает из обоих желудочков в аорту и в меньшем количестве – в лёгочную артерию. Вследствие того, что аорта широкая и смещена вправо, кровь по ней проходит беспрепятственно, поэтому при классической форме тетрады Фалло никогда не бывает недостаточности кровообращения. Из-за большого дефекта межжелудочковой перегородки давление в обоих желудочках одинаковое. Степень гипоксии и тяжесть состояния больных коррелируют со степенью стеноза лёгочной артерии. У пациентов с крайней формой тетрады Фалло кровь в лёгкие поступает через открытый артериальный проток либо через коллатерали (артериовенозные анастомозы и бронхиальные артерии), которые могут развиваться внутриутробно, но чаще - постнатально. Компенсация кровообращения происходит за счет: 1) гипертрофии правого желудочка; 2) функционирования ОАП и/или коллатералей; 3) развития полиглобулии и полицитемии в результате длительной гипоксии;
Цианоз – основной симптом тетрады Фалло. Степень цианоза и время его появления зависит от выраженности стеноза лёгочной артерии. У детей первых дней жизни по признаку цианоза диагностируются только тяжелые формы порока – «крайняя» форма тетрады Фалло. В основном, характерно постепенное развитие цианоза (к 3 мес - 1 году), имеющего различные оттенки (от нежно-голубоватого до «сине-малинового» или «чугунно-синего»): сначала возникает цианоз губ, затем слизистых оболочек, кончиков пальцев, кожи лица, конечностей и туловища. Цианоз нарастает с ростом активности ребенка. Рано развиваются «барабанные палочки» и «часовые стекла». Постоянным признаком является одышка, отмечающаяся в покое и резко возрастающая при малейшей физической нагрузке. Постепенно развивается задержка физического развития. Практически с рождения выслушивается грубый систолический шум вдоль левого края грудины. Грозным клиническим симптомом при тетраде Фалло, обуславливающим тяжесть состояния больных, являются одышечно-цианотические приступы. Возникают они, как правило, в возрасте от 6 до 24 месяцев на фоне анемии. Патогенез возникновения приступа связан с резким спазмом инфундибулярного отдела правого желудочка, в результате чего вся венозная кровь поступает в аорту, вызывая резчайшую гипоксию ЦНС. Насыщение крови кислородом во время приступа падает до 35%. Интенсивность шума при этом резко уменьшается вплоть до полного исчезновения. Ребенок становится беспокойным, выражение лица испуганное, зрачки расширены, одышка и цианоз нарастают, конечности холодные; затем следует потеря сознания, судороги и возможно развитие гипоксической комы и летальный исход. Приступы различны по тяжести и продолжительности (от 10-15 секунд до 2-3 минут). В послеприступном периоде больные длительно остаются вялыми и адинамичными. Иногда отмечается развитие гемипарезов и тяжёлых форм нарушения мозгового кровообращения. К 4-6 годам частота возникновения и интенсивность приступов значительно уменьшается или они исчезают. Связано это с развитием коллатералей, через которые происходит более или менее адекватное кровоснабжение легких.
В зависимости от особенностей клиники выделяют три фазы течения порока:
I фаза – относительного благополучия (от 0 до 6 месяцев), когда состояние пациента относительно удовлетворительное, нет отставания в физическом развитии;
II фаза - одышечно-цианотических приступов (6-24 мес), для которой характерно большое число мозговых осложнений и летальных исходов;
III фаза – переходная, когда клиническая картина порока начинает принимать взрослые черты;
При бледной форме ТФ течение и клинические признаки напоминают таковые при септальных дефектах.
При естественном течении ВПС средняя продолжительность жизни составляет 12-15 лет. Причинами смерти являются одышечно-цианотические приступы, гипоксия, нарушения гемо- и ликвородинамики, тромбозы сосудов головного мозга, инсульты, инфекционный эндокардит.
Хирургическая коррекция больным с ТФ подразделяется на паллиативные операции (наложение подключичного-лёгочного анастомоза) и радикальную коррекцию ВПС. Паллиативная операция позволяет больному выжить и окрепнуть перед проведением радикальной коррекции, которая проводится через 2-3 года. Радикальная операция подразумевает одновременное устранение всех сердечных аномалий и проводится в дошкольном возрасте.
Коарктация аорты(КА) - это врождённое сегментарное сужение аорты в области дуги, перешейка, нижнего грудного или брюшного отделов.
В подавляющем большинстве случаев у грудных детей коарктация располагается на участке от левой подключичной артерии до открытого артериального протока или сразу после него, называемым перешейком аорты.
У плода и новорождённого область перешейка аорты в норме сужена, так как в период внутриутробной жизни через нисходящую аорту поступает всего треть объёма крови, другие две трети проходят через ОАП. Вскоре после закрытия ОАП через перешеек начинает проходить вся кровь, он постепенно расширяется и почти достигает диаметра нисходящей аорты. При наличии патологии область перешейка на том или ином протяжении остаётся суженной. Сужение может иметь вид перетяжки (тогда внутри сосуда обнаруживается мембрана с небольшим отверстием, однако может быть и полный перерыв дуги аорты) или тубулярного сужения на некотором протяжении. По отношению к ОАП коарктация аорты подразделяется следующим образом:
1. Сужение проксимальнее места отхождения ОАП – предуктальная коарктация аорты;
2. Сужение на уровне отхождения ОАП – юкстадуктальная коарктация аорты;
3. Сужение дистальнее отхождения ОАП – постдуктальная коарктация аорты.
Гемодинамика существенно зависит от типа и локализации коарктации, степени сужения, а также от наличия сопутствующих ВПС. При изолированной КА в большом круге кровообращения устанавливаются два режима кровообращения: проксимальнее (артериальная гипертензия) и дистальнее (артериальная гипотензия и дефицит кровотока) места сужения.
При постдуктальной коарктации в случае внутриутробного формирования коллатерального кровообращения течение порока бывает менее тяжелым. Коллатеральный кровоток происходит через подключичные, межреберные, внутренние грудные, лопаточные, эпигастральные и позвоночные артерии, которые со временем расширяются из-за повышенного в них давления. При неадекватном развитии коллатералей существенно повышается артериальное давление до места сужения и кровь под давлением сбрасывается из аорты через ОАП в лёгочную артерию. Величина сброса зависит от градиента между аортой и лёгочной артерией и, как правило, бывает значительной. В ответ на поступление большого дополнительного объёма крови в сосуды малого круга кровообращения, развивается лёгочная гиперволемия и гипертензия.
При предуктальной коарктации аорты направление шунтирования крови будет зависеть от соотношения давлений в нисходящей аорте и лёгочной артерии. При выраженной степени сужения отмечается венозно-артериальный шунт крови, что приводит к появлению дифференцированного цианоза (есть на ногах, но нет на руках).
При сочетании КА с другими ВПС, в частности, с ДМЖП, величина артерио-венозного сброса бывает очень большой, а лёгочная гипертензия развивается быстрее.
Результатом длительных гемодинамических нагрузок является развитие фиброэластоза эндомиокарда левого желудочка, всегда сопровождающегося кардиомегалией, падением сердечного выброса, выраженными признаками гипертрофии левого желудочка и левого предсердия, рефрактерной к лечению сердечной недостаточности.
При постдуктальной локализации КА клиническая картина развивается достаточно быстро – в первые недели жизни. Для детей характерны выраженное беспокойство, одышка, затруднения во вскармливании, развитие гипотрофии. Кожные покровы бледные, с пепельным оттенком (особенно во время приступов беспокойства). Ножки у детей всегда холодные наощупь вследствие дефицита периферического кровотока. Может развиваться деформация грудной клетки по типу «сердечного горба». В легких выслушиваются застойные крепитирующие хрипы, возможно присоединение пневмонии. Сердечный толчок усилен, разлитой. Границы сердца расширены влево и вправо, при фиброэластозе – значительно. При аускультации всегда отмечается тахикардия, иногда – ритм галопа. Шумовая картина неспецифична – чаще всего выслушивается систолический или систоло-диастолический шум ОАП. Может выслушиваться средне- или слабоинтенсивный систолический шум в межлопаточной области. Наиболее специфическим клиническим симптомом, по наличию которого можно заподозрить коарктацию, является снижение пульсации на бедренной артерии. При измерении систолического артериального давления отмечается значительное его повышение в верхней половине туловища. Другими клиническими признаками могут быть симптомы недостаточности кровообращения, как правило, тотальной у маленьких пациентов.
При предуктальной коарктации наряду с вышеуказанными симптомами характерным является наличие дифференцированного цианоза, в большей степени выраженного на ногах.
У детей старшего возраста клиническая картина значительно отличается от таковой у грудных пациентов. Как правило, дети развиваются нормально. Порок выявляется случайно (чаще в школьном возрасте) при обнаружении повышенного артериального давления. Характерен внешний вид таких детей с развитой верхней половиной туловища и астеническим телосложением нижней половины тела.
 На этапе первичной адаптации отмечается высокая смертность детей вследствие тяжелой сердечной недостаточности и присоединения пневмоний. В дальнейшем состояние больных стабилизируется (за счёт развития коллатерального кровообращения и гипертрофии миокарда, закрытия ОАП) и они доживают, в среднем, до 30-35 лет. Основными осложнениями у взрослых являются расслаивающаяся аневризма и разрыв аорты, тяжелые инсульты и инфекционный эндокардит.
На этапе первичной адаптации отмечается высокая смертность детей вследствие тяжелой сердечной недостаточности и присоединения пневмоний. В дальнейшем состояние больных стабилизируется (за счёт развития коллатерального кровообращения и гипертрофии миокарда, закрытия ОАП) и они доживают, в среднем, до 30-35 лет. Основными осложнениями у взрослых являются расслаивающаяся аневризма и разрыв аорты, тяжелые инсульты и инфекционный эндокардит.
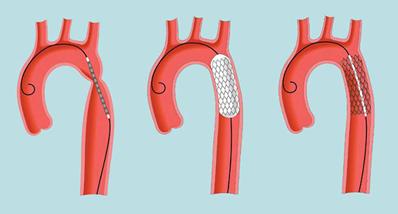
 Хирургическая коррекция заключается в иссечении участка сужения аорты и соединения иссечённых концов «конец в конец», «бок в бок», «конец в бок», либо последующей истмопластики. У детей с коарктацией по типу внутренней мембраны может применяться операция баллонной ангиопластики (рис.8).
Хирургическая коррекция заключается в иссечении участка сужения аорты и соединения иссечённых концов «конец в конец», «бок в бок», «конец в бок», либо последующей истмопластики. У детей с коарктацией по типу внутренней мембраны может применяться операция баллонной ангиопластики (рис.8).