ВВЕДЕНИЕ

Определение и характеристика функции белков в токсоплазме был в центре обширных исследований этого паразит, так как наличие методов, которые сделали возможным изучение отдельных генов и молекул. Механизм вторжения хост-клетки, структуры и состава апикальных органелл, конверсии стадии, и уклонение от хозяина иммунной защиты были, в частности, являются важными и повторяющиеся темы. Наше понимание экспрессии генов в контексте этих фундаментальных проблем было нанято секвенирование генома Gondii Т. и последующего развитием сложных генетических инструментов для изучения этого паразита.
Белки являются конечным продуктом генов, и это в конечном счете действия белков, а не генов, которые играют центральную роль в фундаментальных процессах паразитизма, которые мы хотим понять. До появления протеомики, крупномасштабное исследование белков не упало значительно отстает от генов. Недавнее развитие протеомическим методов Т. гондий и наличие последовательности генома для подкрепления протеомный базы данных в настоящее время представляют собой беспрецедентную возможность охарактеризовать состав и функции белков в паразита.
Для того, чтобы понять эволюцию протеомических шпильки-х года в токсоплазмах, полезно кратко взглянуть на развитии транскрипционного анализа для этого паразита. Microarrays обеспечивают захватывающую возможность изучения транскрипционных моделей не
Токсоплазма. Модельные Apicomplexan-перспективы и методы, под редакцией Weiss & Kim
ISBN-13: 978-0-12-369542-0
ISBN-10: 0-12-369542-2
Copyright © 2007 Elsevier Ltd.
Все права воспроизведения в любой форме зарезервированы.

542 ПРОТЕОМИКА OF токсоплазма
Горстка но тысячи генов в любое время в различных биологических контекстах. Ряд крупномасштабных исследований экспрессии транскрипции гена с использованием микрочипов уже предпринимались для T. гондий (Blader и др., 2001; Клири и др., 2002; Matrajt и др., 2002; Сибли и др., 2002; Singh и др., 2002). Однако транскрипционное исследование имеет ряд недостатков. Существует менее чем предсказуемое соотношение между геном transcrip-ции и экспрессия белка, хотя недавнюю работа с использованием биосинтетической маркировки РНКА с урацилом фосфорибозилтрансферазой, чтобы позволить микрочип анализа синтеза мРНКа и распада в T.gondii, может пройти определенный путь для решения этой проблемы (Клиря и др., 2005). Кроме того, активность белки зависит не только от его избытка, но и от его состояния активации, часто опосредовано через пост-трансляционное событие, такие как фосфорилирования. Наконец, активность белки также зависит от взаимодействия с другими белками, так что идентифицируемая-фикация и пониманием природы этих взаимодействий имеет жизненно важное значение при изучении функции белка. К сожалению, микрочип на основе транскрипционных исследований дают небольшую помощь при решении этих вопросов. Протеом, по определению, содержит все белки, выраженные во всех их различных состояниях активации и модификации. Протеомика или, возможно, более целесообразно, функциональные протеомики, таким образом, имеет потенциал, чтобы ликвидировать разрыв между нашим знанием генома и функциональных биологических процессов, присущих организму. так что идентифицируемая-фикация и понимание природы этих взаиморасчетов действий имеет жизненно важное значение при изучении функции белка. К сожалению, микрочип на основе транскрипционных исследований дают небольшую помощь при решении этих вопросов. Протеом, по определению, содержит все белки, выраженные во всех их различных состояниях активации и модификации. Протеомика или, возможно, более целесообразно, функциональные протеомики, таким образом, имеет потенциал, чтобы ликвидировать разрыв между нашим знанием генома и функциональных биологических процессов, присущих организму. так что идентифицируемая-фикация и понимание природы этих взаиморасчетов действий имеет жизненно важное значение при изучении функции белка. К сожалению, микрочип на основе транскрипционных исследований дают небольшую помощь при решении этих вопросов. Протеом, по определению, содержит все белки, выраженные во всех их различных состояниях активации и модификации. Протеомика или, возможно, более целесообразно, функциональные протеомики, таким образом, имеет потенциал, чтобы ликвидировать разрыв между нашим знанием генома и функциональных биологических процессов, присущих организму. содержит все выраженные белки во всех их различных состояниях активации и модификации. Протеомика или, возможно, более целесообразно, функциональные протеомики, таким образом, имеет потенциал, чтобы ликвидировать разрыв между нашим знанием генома и функциональных биологических процессов, присущих организму. содержит все выраженные белки во всех их различных состояниях активации и модификации. Протеомика или, возможно, более целесообразно, функциональные протеомики, таким образом, имеет потенциал, чтобы ликвидировать разрыв между нашим знанием генома и функциональных биологических процессов, присущих организму.
Переход от генома Транскрипта, а затем протеома является не только возрастающей сложностью, но и повышения нестабильности. Протеомов являются весьма динамическими объектами. Даже если бы это было технически возможно «карты» по-настоящему полный протеома для токсоплазмов (и в настоящее время это не так), это протеом будет характерна только для точных биологических условий, в которых были подготовлены паразиты для анализа. Это будет включать стадии их жизни, точку в их клеточном цикле, были ли производные паразиты в пробирке или в естественных условиях, и т.д. Таким образом, сложность протеомов, в то время как Techni-ческах сложный, обеспечивает индикацию потенциальной мощности анализа протеомики. Так как простейшие, такие как Toxoplasma являются сложным microor-ganisms, любой анализа протеого который способен
отражаем поэтому эта сложность имеет потенциал, чтобы обеспечить ключ к пониманию того, как этот паразит функции и взаимодействует с окружающей средой.
Хотя крупномасштабные микрочипов и протеомики исследования дают новые захватывающие возможности, они также привлекли некоторую критику. Многие ранние проблемы как микрочип и протеомики были из-за неадекватные инструменты биоинформатики для обработки беспрецедентного количества сложных данных, что этот типа анализа производства. Хотя многое еще предстоит сделать, в последние годы сущест-косяка успехи были достигнуты в разработке мощных инструментов биоинформатики для обработки этих данных (Hancock и др., 2002; Haley-Висенте и Edwards, 2003; Taylor и др., 2003; Джонс и др., 2004;. Хамади и др, 2005; Кремер и др, 2005;. Souchelnytskyi, 2005). Другие критические замечания имеют более философскую основу. До так называемого «пост-геномного эпохи», типичный обычный molecu-лар исследование Toxoplasma, возможно, начали с идентификацией одного гена или белка, связанного с конкретным событием биологии, таких как вторжение или стадии конверсии. Этот ген, как правило, быть секвенирован, клонирован, и повторно выражен, чтобы обеспечить локализацию и другие функциональные данные; его структура гена, возможно, была нанесена на карту Южного блоттинга, его транскрипционная экспрессию с помощью Нозерн-блоттинга, и экспрессии белка, который он кодирует помощью Вестерн-блоттинга. Гомологичные гены, возможно, затем были запрошены с использованием вырожденных праймеров для ПЦР, и процесс повторяется. Следовательно, большое количество подробной информации была собрана об очень небольшом количестве генов, часто основаны вокруг одной гипотезы. Многие прекрасные исследования, основанные на вышеуказанную гипотезу «управляемая» шаблон обеспечили заземление для наших настоящего под-стояний Т. гондий; Однако этот процесс идет медленно и не использует нынешний объем данных последовательностей генома в наиболее эффективном способе. В противоположность этому, микрочипов и протеомики эксперименты дают изначально более низкого класса информации, а не на несколько, а на сотни или даже тысячи генов. Кроме того, типы генов и белков, чтобы исследовать, прежде чем эксперимент, предварительно не выбрано (следовательно, иногда привлекая уничижительное описание «рыболовных экспериментов»). а не на несколько, а на сотни или даже тысячи генов. Кроме того, типы генов и белков, чтобы исследовать, прежде чем эксперимент, предварительно не выбрано (следовательно, иногда привлекая уничижительное описание «рыболовных экспериментов»). а не на несколько, а на сотни или даже тысячи генов. Кроме того, типы генов и белков, чтобы исследовать, прежде чем эксперимент, предварительно не выбрано (следовательно, иногда привлекая уничижительное описание «рыболовных экспериментов»).
| ОСНОВЫПРОТЕОМИКИ |
В самом деле, крупномасштабные микрочип и протеомика эксперименты были специально разработаны, чтобы не предвосхищают результаты анализа путем выявления-кий в несмещенных генах пути и белках, которые связаны с конкретными биологическими событиями. Эти эксперименты, таким образом, важную роль в формировании гипотезы, а не обязательно всегда быть гипотеза тестирования. Масштабные микрочип и протеомика эксперименты, следовательно, не в конфликте с так называемыми гипотезами экспериментов, но должна быть партнерами в рамках итеративного процесса разработки гипотез и тестирования.
В этой главе мы рассмотрим современное состояние протеомики анализа Toxoplasma и обсудить, как эти данные были использованы для улучшения нашего понимания биологии этого паразита. Мы смотрим на конкретных примерах того, как данные протеомики и подтвердили предыдущие гипотезы и показали неожиданные результаты, которые были бы трудно предсказать без принятия протеомики подхода. Разработанный в конце 1990-х годов, протеомики является относительно молодой областью, хотя почти с момента ее создания протеомики эксплуатировалось тех, кто заинтересован в биологии токсоплазмы. В таком быстро развиваться-Инг субъекта, это неизбежно, что деталь любого обзора будет устаревать практически сразу. Однако цель этой главы является не только представить обновленную информацию о нынешнем состоянии исследований,
ОСНОВЫПРОТЕОМИКИ
Есть много прекрасных общие обзоры по протеомики в литературе (Aebersold и Mann, 2003; Morrison и др, 2003;. Gorg и др., 2004; Johnson и др., 2004; Yates, 2004; Bradshaw и Бёрлингейм, 2005; Dhingra и др, 2005;. Лейн, 2005; Филлипс и Bogyo, 2005); эти и другие должны быть переданы для более тщательного ознакомления с предметом. Однако, прежде чем дело конкретно с протеомики исследований Toxoplasma мы будем
Кратко рассмотрим некоторые из основных принципов этой относительно новой области техники.
Масс-спектрометрический анализ (МС) белки существенно заменить классические методы белок микро-последовательность, такие как Эдман. МС позволяет измерения массы ионов, полученных из молекул, и способен образовывать, SEPA-рейтинге, и выявления молекулярных ионов на основе их массы к заряду рациона (m / z). Разработать Единение двух методов ионизации в поздних 1980-х из MS обычно доступны для биологических исследователей. Они были матрично-активированная лазерная десорбция / ионизация масс-спектрометрии (MALDI-MS), и электро-спрей ионизации тандемной масс-спектрометрии (ESI-МС / МС). Протеомики по существу влечет за собой согласование двух наборов данных, один экспери-ментального и один виртуального, чтобы обеспечить быстрое identifica Тиона белков. Общая схема для протеомики эксперимента показана на рисунке 20.1. Experimen-тал набор данных состоит из данных МС, который в простейшем виде содержат точные массы пептидов, полученных из белков переваренных ферментом, таким как трипсин. Измерено с помощью MALDI-MS, отдельные массы каждого пептида приводят к массовому отпечатка пальца уникальный пептид (PMF). Альтернативно, пептиды могут быть фрагментированы с помощью ESI-MS / MS для получения масс-спектрометрии, или тандем MS / MS, данные. Еще раз уникальный отпечаток пальца получается, на этот раз для каждого отдельного пептида. В обоих случаях, экспериментальные данные MS затем могут быть сопоставлены с использованием алгоритмов к соответствующей базе данных массового виртуального пептида. Виртуальные массовые базы данных зависят от наличия значительного количества последовательности генома, или данных EST, и выводятся из сдвигов всех possi-BLE открытых рамок считывания в виде белковых последовательности, или из моделей прогнозирования белка.
Растворимые и нерастворимые протеомов
Успешное применение протеомики в решающей степени зависит от обогащения и разделения
544 ПРОТЕОМИКА OF токсоплазма

| Виртуальный протеом | массовая база Пептид | ||||

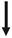

идентификация белка

| Реальный протеом | Протеин данных MS | ||||

Рисунок 20.1 Общая схема для протеомики эксперимента. В протеомики, идентификация белков облегчается за счет согласования двух наборов данных, один экспериментальный (снизу) и одной виртуальной (вверху). Parasite белки могут быть разделены с помощью различных методов, таких, как двухмерного электрофореза, а отдельные белки затем анализировали с помощью масс-спектрометрии (МС), чтобы произвести экспериментальный набор данных, состоящий из либо пептидных массовых отпечатков пальцев (MALDI-MS), или пептид фрагментации данных (ESI-MS / MS). Экспериментальные данные MS затем согласованы с использованием соответствующих алгоритмов с соответствующей базой данных массовых виртуального пептида, полученных из последовательности генома, или данных EST и содержащих переводов всех possi-BLE открытых рамок считывания в белках последовательности. Так как завершение секвенирования генома для проекта Toxoplasma эти гипотетические базы данных белковых могут быть легко изготовлены из ToxoDB, что дает возможность точного совпадения с экспериментальными данными MS и быстрой идентификации T. гондий белков. Эта цифра воспроизводится в цвете в разделе Цвет пластины.
белки под следствием до анализа MS.
и образцы, содержащие белок переваривается
Метод выбора для разделения белков
и анализировали с помощью МСА для идентификации каждого белок пятна.
зависит от природы белков: как правило,
К сожалению, некоторые белки, в частности, гидрофобные
либо двумерный электрофорез (2-DE)
белки и те, в крайних молекулярном
или на основе не-гель методы, такие как жидкий хроматографе
вес, как известно, трудно решить с помощью
matography (ЖХ) используются. 2-DE особенно
2-DE. Очень низкие белки обилие также
подходит для анализа растворимых протеомов, и
трудно обнаружить 2-DE.
производит с высоким разрешением двухмерных карт
Альтернативой разделения белков 2-DE является
белковых компонентов, которые позволяют относительное
провести двумерный разделение белка
Уровень экспрессии стационарного белка, чтобы быть
с использованием комбинации либо одномерным
По оценкам, а также потенциальной идентификации
SDS-PAGE с последующим нано-жидкостной хроматографический
пост-трансляционной модификации. Индивидуальный
PHY и МС, или два последовательных жидкостной хроматографии
белковые пятна, то могут быть выбраны из геля,
разделения с последующим МСОМ. В последнем подходе,
| КОТОРАЯ Протеый? Протеомы и суб-протеомы Т. гондий |
также известный как многомерную технологию белок identifica-ница (MudPIT), белки перевариваются с помощью трипсина, и пептиды отделены первым использованием сильного катионита и второй обращенно-фазовой LC. Пептиды, которые элюированные из второй колонны LC подают непосредственно в ионно-ловушке масс-спектрометр ESI, где они ионизируются, масса выбраны, и фрагментированы. Фрагментированные пептиды затем согласованы с белками в базе данных с использованием соответствующих алгоритмов. MudPIT был использован частично разрешить протеомы ряда микробов, в том числе дрожжи, где 1484 белки и пептиды 5540 были решены и идентифицированы из Saccharomyces Cere-visiae (Washburn и др., 2001). Значительно эти белки содержали низкое обилие белков, такие как факторы транскрипции и киназы, а также белки с крайностями PI и молекулярной массой, и мембранные белки обычно не разрешаются 2-DE. Оба MudPIT и одномерным SDS-PAGE с последующим нано-LC были использованы для анализа протеомов протозойных паразитов, в том числе малярийного плазмодия (Florens, и др., 2002;. Lasonder и др, 2002) и Trypanosoma Тгурапозота (ПАБК и др., 2004). Анализ тахизоитов T.gondii, по MudPIT в настоящее время ведется (Wastling, неопубликованные данные).
Во-первых, анализ MudPIT может показаться, что очевидной заменой для протеомики 2-DE на основе, как она обходит многих своих ограничений, присущих и предлагает относительно процедуры «невмешательстве», который может быстро получать значительные объемы данных. Тем не менее, простота MudPIT происходит за счет потери информации о белках, которые анализируются. Одним из преимущественных признаков разделения белков 2-DE является то, что она раскрывает сложность протеома таким образом, что MudPIT в данный момент не может. Так, например, 2-ДЕ протеомный анализ Т. гондий (Cohen и др., 2002) и других паразитов (Jones и др., 2006) показывает, что отдельные белки часто представлены более чем в одном месте, гель. Эти дополнительные пятна не являются артефактами, но представляют собой потенциально важные функциональные особенности белков, такие как альтернативное событие сплайсинга, или пост-трансляционной модификации, как фосфорилирования. Кроме того, в то время как относительное обилие белков, представленных на 2-DE гель
может быть воспроизводимо по сравнению с использованием сравнительных подходов гель (т.е. Dige), MudPIT, как известно, не количественный характер. Это последнее свойство может представлять особые трудности при анализе обогащенных суб-протеомов, поскольку различие между суб-протеомом под следствием и незначительные примеси могут быть весьма проблемно-ATIC (Sam-Yellowe и др., 2004).